Article Text

Abstract
Objectives A pharmacy Central Intravenous Additives Service (CIVAS) provides ready to use injectable medicines. However, manipulation of a licensed injectable medicine may significantly alter the stability of drug(s) in the final product. The aim of this study was to develop a stability indicating assay for CIVAS produced dobutamine 500 mg in 50 ml dextrose 1% (w/v) prefilled syringes, and to allocate a suitable shelf life.
Methods A stability indicating high performance liquid chromatography (HPLC) assay was established for dobutamine. The stability of dobutamine prefilled syringes was evaluated under storage conditions of 4°C (protected from light), room temperature (protected from light), room temperature (exposed to light) and 40°C (protected from light) at various time points (up to 42 days).
Results An HPLC method employing a Hypersil column, mobile phase (pH=4.0) consisting of 82:12:6 (v/v/v) 0.05 M KH2PO4:acetonitrile:methanol plus 0.3% (v/v) triethylamine with UV detection at λ=280 nm was specific for dobutamine. Under different storage conditions only samples stored at 40°C showed greater than 5% degradation (5.08%) at 42 days and had the shortest T95% based on this criterion (44.6 days compared with 111.4 days for 4°C). Exposure to light also reduced dobutamine stability. Discolouration on storage was the limiting factor in shelf life allocation, even when dobutamine remained within 5% of the initial concentration.
Conclusions A stability indicating HPLC assay for dobutamine was developed. The shelf life recommended for the CIVAS product was 42 days at 4°C and 35 days at room temperature when protected from light.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ejhp.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Dobutamine is an inotropic synthetic catecholamine which acts on adrenergic sympathetic receptors and is used to treat the majority of patients with cardiogenic shock at Guy's and St Thomas' NHS Foundation Trust (GSTFT). The drug is a racemic mixture of (+)-dobutamine, which has strong β agonist and some α antagonist activity, and (−)-dobutamine which is an α agonist and weak β2 antagonist. The overall therapeutic effect of a racemic mixture of dobutamine is a blend of these activities. As dobutamine has a short systemic half life (∼2 min) and is not bioavailable orally,1 it is administered as an intravenous infusion. On the hospital wards at GSTFT, 40 ml of concentrated dobutamine hydrochloride solution (12.5 mg/ml) are diluted with dextrose 5% (w/v) solution to a total volume of 50 ml; this is administered using a syringe pump. This method of preparation is time consuming and introduces risks to patient safety due to the potential for microbial contamination and calculation error.
Following National Patient Safety Agency alert 20,2 the GSTFT pharmacy identified dobutamine 500 mg in 50 ml infusion as one of the higher risk injectable medicines in use in the Trust. To manage this risk, dobutamine 500 mg in 50 ml of dextrose 1% (w/v) was selected for provision in prefilled syringes as part of the existing pharmacy Central Intravenous Additives Service (CIVAS). Under CIVAS, medicines are prepared in the controlled environment of a pharmacy aseptic unit and presented in ready to use form for administration on the ward.3 This service evolved as a risk management approach to avoid the risks associated with ward based preparation of intravenous additives—for example, microbial contamination, formulation instability and procedural errors. The CIVAS approach is a well established part of hospital pharmacy services in the UK.
The preparation of dobutamine 500 mg in 50 ml of dextrose 1% (w/v) dose units was initiated using a ‘multidispensing’ model whereby licensed dobutamine injection concentrate was diluted to produce individual syringes in small batches of 10 syringes. Using this procedure, quality control (QC) testing of drug content is of limited value as no unit is representative of the batch and these CIVAS product batches were released by the aseptic unit for administration without the standard final analytical QC release test for GMP manufactured products. Final release was based on the reconciliation of the licensed materials used, the preparation of the products by accredited staff and a review of the final completed batch record sheet by the quality assurance department.
When the demand for dobutamine syringes increased, the ‘multidispensing’ method was considered inappropriate for scale up of batch size due to insufficient assurance of the drug content of the final product. A production method was devised whereby dobutamine 10 mg/ml in dextrose 1% (w/v) was prepared aseptically as a bulk solution in 6 l infusion bags and then transferred aseptically into syringes using a semiautomated pump. This method required the development of an assay for dobutamine to: (1) evaluate the uniformity of mixing achieved by the manufacturing process, (2) verify the content of dobutamine in the final product by QC testing and (3) perform stability indicating studies to determine a suitable shelf life for the dobutamine syringes.
There are limited published data on the chemical stability of dobutamine. It has been reported that the drug is rapidly broken down under basic conditions to form brown coloured polymers via complex chemical pathways.4 Although dobutamine is stable in acidic conditions, it is susceptible to degradation in light.4 However, it has been noted that dobutamine 5 mg/ml in dextrose 5% (w/v) is stable in disposable 60 ml plastic syringes for 24 h at 25°C.5 Other studies have also reported the chemical stability of dobutamine injections of various concentrations under various storage conditions,6 7 but do not provide sufficient or relevant data on which to assign a shelf life for the CIVAS product. High performance liquid chromatography (HPLC) has also been used to demonstrate stability based on percentage drug remaining over 24 h when dobutamine was added to peritoneal dialysis solutions at final concentrations of 2.5, 5.0 and 7.5 mg/ml and stored at temperatures of 4°C, 26°C and 37°C.8 Further HPLC methods have been described for the separation and quantification of dobutamine in plasma.9 10 However, no stability indicating assay for dobutamine has been reported to date that satisfies all of the criteria required for the quality assurance/QC purposes described herein.
The standard for stability testing of drug products (ICH guideline Q1A R2) defines a significant change to a drug product as: (1) a 5% change in assay from its initial value (the time taken for this change to occur is T95%); (2) appearance of any degradation product exceeding its acceptance criterion; and (3) failure to meet the acceptance criteria for appearance, physical attributes and functionality.11 We report a simple HPLC assay for dobutamine 500 mg in 50 ml of dextrose 1% (w/v) solution that is suitable for use in regular QC activity and can also be used as a stability indicating assay to determine T95% as part of the process of assigning product shelf life. The assay was used to verify the fitness of new manufacturing procedures and determine the stability of dobutamine in prefilled syringes under a variety of storage conditions.
Materials and methods
Dobutamine hydrochloride reference powder was obtained from USP (Rockville, USA) and was used without further purification. Dobutamine hydrochloride concentrate (12.5 mg/ml; 20 ml vials) injections were obtained from Phoenix Pharma (Gloucester, UK); dextrose 5% (w/v) solution was obtained from Maco Pharma (Middlesex, UK) and potassium dihydrogen orthophosphate (KH2PO4) from Fisher Scientific (Loughborough, UK). Dopamine, anisaldehyde and 4-(4-hydroxy phenyl)-2-butanone were obtained from Fluka (UK) while methanol, triethylamine and acetonitrile were obtained from Sigma-Aldrich (Dorset, UK). Becton Dickinson 50 ml syringes were obtained from Becton Dickinson (Oxford, UK). All reagents used were analytical grade. A refrigerator was used to maintain a storage temperature of 4.0 (±1)°C. Room temperature was monitored throughout the period of experimentation and was found to have a midday value of 20.0 (±4)°C. A thermostatically controlled incubator was used to maintain a storage temperature of 40.0 (±2)°C.
HPLC analysis
The Merck–Hitachi HPLC system used in the present study consisted of a pump (L-6200A), ultraviolet (UV) light detector (L-400) and integrator (D-2500). The system was fitted with a 20 µl injection loop and separation was achieved using a HiChrom Hypersil octadecylsilane column (100 mm×4.5 mm×5 µm) with 10% carbon loading. UV studies were carried out on a calibrated Varian Cary |1| UV visible spectrophotometer. The isocratic flow rate was 1.0 ml/min and the UV detector was set at a wavelength of 280 nm.
The HPLC mobile phase consisted of 82:12:6 (v/v/v) of 0.05 M KH2PO4:acetonitrile:methanol plus 0.3% (v/v) triethylamine and was adjusted to pH 4.0 (±0.1) using phosphoric acid. The mobile phase solution was degassed by filtration through a 0.45 µm nylon membrane followed by ultrasonication for 1 h. Standards used in the calibration of the HPLC method were prepared by dissolving dobutamine hydrochloride powder in the HPLC mobile phase solution with the aid of ultrasonication. QC standards were obtained from a stock solution prepared by dissolving 100 mg of dobutamine hydrochloride powder in 10 ml of the HPLC mobile phase solution, producing a concentration similar to that of the CIVAS product (10 mg/ml). Concentrations of 0.25, 0.50, 0.75, 1.00, 1.25 and 1.50 mg/ml of dobutamine were used to establish linearity. Concentrations of 0.25, 0.75 and 1.00 mg/ml of dobutamine were prepared by dilution of the stock solution by mobile phase and these were used for method validation (specificity, repeatability, range, precision and accuracy) according to ICH guidelines. Specificity was also determined by analysing mixtures from the accelerated degradation studies. Known concentrations of dobutamine were spiked into these mixtures and any increase in the area under the dobutamine peak was measured. Inter- and intra-day accuracy and precision were evaluated by analysis of independent samples. Repeatability was assessed via multiple injections of the same standard solution. All samples were measured in triplicate except in the validation of method repeatability where 10 replicates were measured.
Accelerated degradation studies
Accelerated degradation studies were conducted using dobutamine solution under various conditions. Decomposition of dobutamine under acidic conditions (0.1, 1 and 2 M HCl) was induced at 60°C for 72 h. Similarly, dobutamine was exposed to alkaline conditions of 0.1 and 1 M NaOH (at 60°C for 12 h), 3 M NaOH (room temperature for 72 h) and 5 M NaOH (room temperature for 3 weeks). Decomposition under neutral conditions (phosphate buffer; pH 7) at 60°C for 72 h was also conducted. The effect of oxidative conditions was studied by exposing the drug to 30% H2O2 at 60°C for 72 h. For photostability studies, two samples were prepared in distilled water; one was placed at room temperature protected from light and the other was exposed to light for 1 week.
Stability of prefilled syringes
For stability testing, a batch of dobutamine 50 ml prefilled syringes was manufactured in the aseptic unit at GSTFT. For the purpose of this study, the contents of two standard CIVAS 50 ml syringes were mixed and 29 samples, containing 3 ml of dobutamine solution each, were prepared aseptically to avoid microbiologically assisted degradation. The 3 ml samples were prepared in 50 ml syringes to match the packaging and storage conditions employed for the standard dobutamine in dextrose product regularly manufactured at the Trust.
In the present study, four different storage conditions were investigated. Excluding the sample analysed at time zero, the remaining 28 syringes were randomly divided into 4×7 syringes and each group was stored at 4°C protected from light, room temperature protected from light, room temperature in light and 40°C protected from light. Over the course of the study (t=0, 3, 7, 14, 21, 28, 35 and 42 days), samples were stored in a single location with minimal manipulation and movement. On each day of analysis, one syringe was taken from each of the four groups, diluted 1 in 10 with mobile phase and analysed by HPLC in triplicate.
Prediction of T95%
The degradation coefficients or rate constants (k; days−1) were determined from the slope of the plot of the log of drug remaining versus time (days) at each of the conditions studied (room temperature was assigned as 22°C). The expiry date (t; days), based on a limit of 5% degradation, was calculated from the degradation coefficient (k) using equation 1:
t=2.303/k.log (Co/C) Equation 1
where Co is the initial percentage of drug at time zero (100%) and C is the percentage of drug remaining at time, t (95%).
An Arrhenius plot (equation 2) was constructed for samples protected from light to estimate stability at this condition at any given temperature:
k=Ae-Ea/RT Equation 2
where k is the rate constant, A is the pre-exponential factor, Ea is the activation energy, R is the gas constant and T is absolute temperature in Kelvin (K).
Statistics
All values are expressed as mean±SD. Statistical evaluation of the data was performed with SPSS (V.15.0; SPSS, Chicago, Illinois, USA). Data were compared using a t test. In all cases, a difference was considered significant at p≤0.05.
Results
HPLC method validation
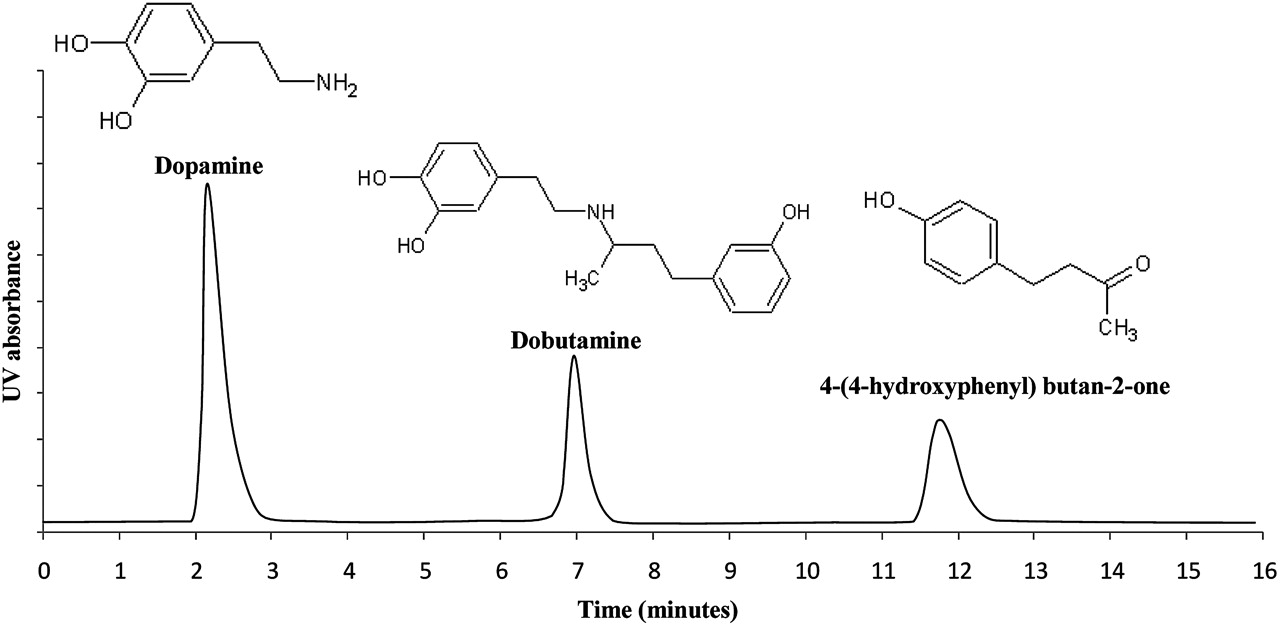
The HPLC method was highly specific for dobutamine, providing good peak resolution from the common impurities dopamine and 4-(4-hydroxyphenyl) butan-2-one (figure 1) and from the products of the stressed samples. Heating of dobutamine in acidic conditions produced no significant degradation (t test: p>0.05). In contrast, dobutamine was highly susceptible to oxidation under alkaline conditions, forming a red/brown product. In peroxide solution at neutral pH, dobutamine was stable with no loss of drug or additional chromatographic peaks arising after 7 days. Dobutamine was sensitive to photodegradation with the appearance of additional peaks in the HPLC chromatograms following exposure to light.

A representative high performance liquid chromatography chromatogram obtained using a mixture of dopamine, dobutamine and 4-(4-hydroxyphenyl) butan-2-one.
Dobutamine had a retention time of approximately 7 min (figure 1). Repeatability testing indicated a CV of 0.31% for 10 replicates. The method was linear over the range 0.25–1.50 mg/ml with a correlation coefficient R2=0.9999. The limits of detection and quantification were calculated to be 0.20 and 0.94 µg/ml, respectively. Intra-day and inter-day accuracy for concentrations in the linear range were acceptable (intra-day, n=6, mean deviation <0.76%; inter-day, n=8, mean deviation <1.18%). Precision was acceptable with intra-day and inter-day values for relative SD of <0.86% (n=6) and <0.90% (n=8), respectively.
Stability of dobutamine containing CIVAS syringes
The stability of dobutamine was dependant on the storage conditions employed (table 1, figure 2). The dobutamine concentration in the syringe assayed immediately postpreparation was within specification: 0.9971±0.0031 mg/ml. The concentration of dobutamine in CIVAS syringes showed a progressive reduction over the storage period. Loss of dobutamine over the 42 day duration of the study remained within the <5% change in concentration limit set for this study for all storage conditions except for storage at 40°C where the loss of dobutamine was 5.08% on day 42 (table 1). The presence of light was shown to contribute significantly (t test: p<0.01) to dobutamine degradation compared with storage in the absence of light. It was noted that some discolouration was observed in samples stored at 40°C in the absence of light (day 35), room temperature in the absence of light (day 42) and room temperature in the presence of light (day 28), even when dobutamine remained >95% of the initial concentration, although no quantifiable degradation peaks (>10 times the baseline noise) were observed.
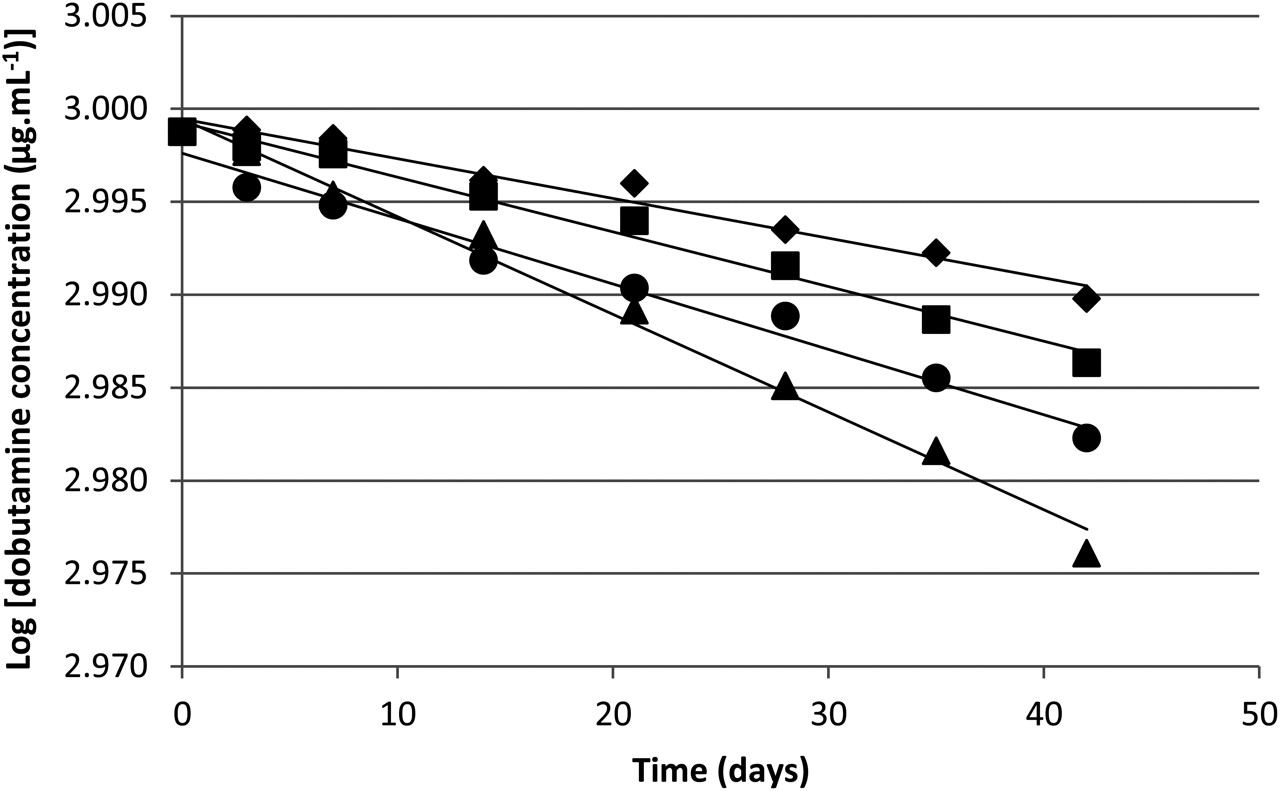
First order kinetic demonstrated by the logarithm of the dobutamine concentration in CIVAS syringes as a function of time, where (♦) represents 4°C (protected from light), (■) represents room temperature (protected from light), (▲) represents 40°C (protected from light) and (●) represents room temperature (exposed to light). Regression analysis was used to obtain the best fit; linear equations and their correlation coefficients are provided in table 2.
Stability of dobutamine 500 mg in 50 ml syringes stored under different conditions of temperature, with and without exposure to light
Predicted T95%
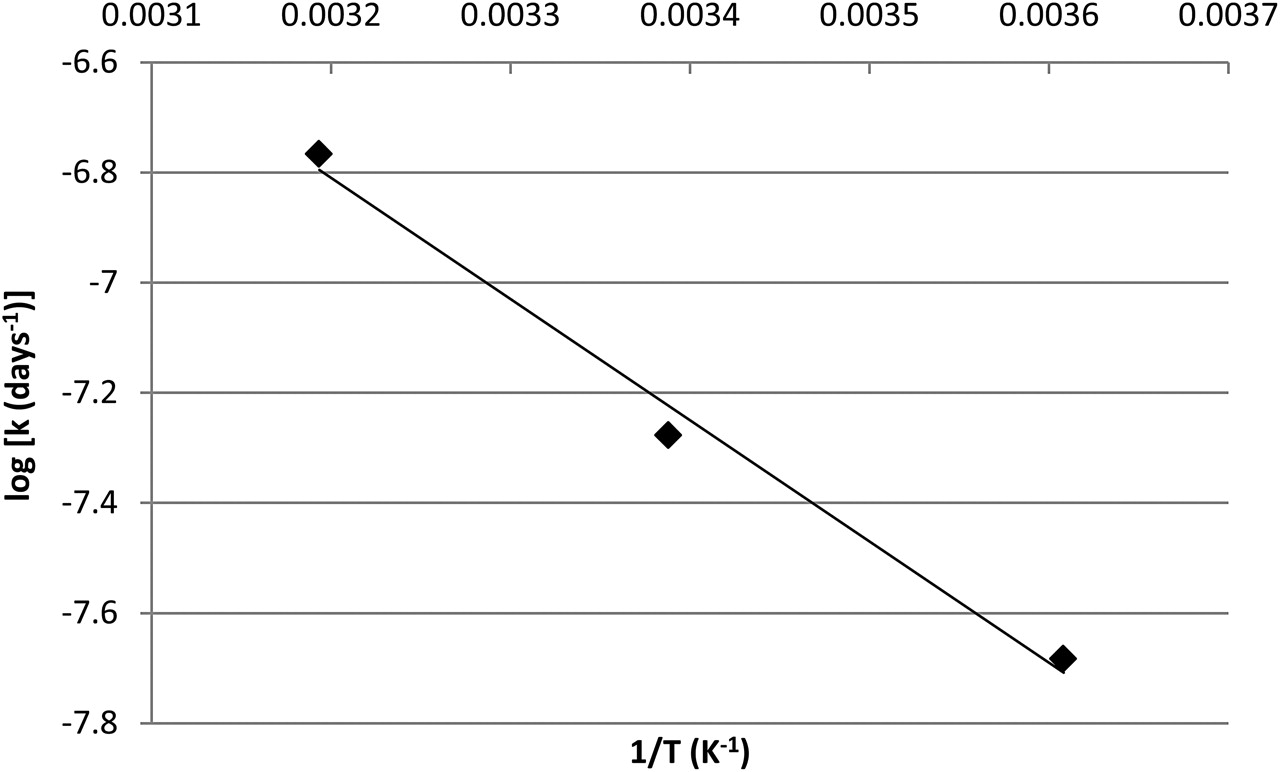
First order reaction kinetics were demonstrated by the linearity of the log of drug remaining versus time (figure 2, table 2). From these data the degradation coefficient and the calculated T95% of dobutamine syringes, based on 5% reduction in drug concentration under the storage conditions tested, were determined (table 2). T95% under the various test conditions varied from 44.6 to 111.4 days, with the lowest T95% when the samples were exposed to 40°C, and the greatest T95% at 4°C and protected from light. Construction of an Arrhenius plot for samples stored protected from light verified the first order reaction kinetics (figure 3). The Arrhenius plot showed a good linear correlation (R2=0.9897) and can be used to calculate the T95% of dobutamine syringes stored protected from light at any given temperature.

{kind=link}
{kind=link}
{kind=link}
An Arrhenius plot demonstrating a linear correlation between the logarithm of the degradation coefficient (days−1) for dobutamine syringes stored protected from light and the inverse of temperature (y=−2201.1x+0.2338; R2=09897). Application of the Arrhenius equation (equation 2) allows the shelf life to be determined for the product stored protected from light for any given temperature.
Linear regression equation, degradation coefficient and calculated T95% of dobutamine in Central Intravenous Additives Service syringes based on 5% loss when stored under various experimental conditions
Discussion
Stability studies are time consuming to perform and infrequently undertaken for CIVAS products. However, a drug may be exposed to multiple chemical and physical environments as part of a CIVAS manufacturing process, which may result in loss of drug. Manipulation of drug compounds, mixing with other active/excipient components and storage in a new container may significantly alter the stability of drug(s) in the final CIVAS product. This means that product stability cannot be inferred unequivocally from published stability data for the drug in the original formulations/system(s). Thus the CIVAS system, which evolved to avoid the risks associated with the ward based preparation of doses for intravenous administration, introduces an element of uncertainty regarding the assurance of product quality.
Drug stability can be verified using stability indicating assays. Although several methods have been reported for the analysis of dobutamine stability,4,–,8 12,–,14 none of these methods reported the resolution required of a stability indicating method when degraded dobutamine samples were analysed. The method reported by Bishara and Long4 provided better separation compared with the other methods evaluated, but the retention time was variable and the dobutamine peak was frequently overlapped by the peaks of degradants. Within our laboratory, a HPLC method was developed that provides good separation of dobutamine and its degradation products using a mobile phase containing 0.05 M KH2PO4, acetonitrile and methanol. The addition of triethylamine improved the reproducibility of the retention time and peak shape. Dobutamine eluted approximately midway (∼7 min) through the run time in a ‘window’ with a stable baseline in which no breakdown product eluted, including all samples subjected to the forced degradation studies. Based on the separation achieved under normal and accelerated degradation studies, the method was considered a dobutamine specific, stability indicating assay, as defined by Bakshi and coworkers.15
Dobutamine is reported to be stable under acidic conditions.4 16 Similar findings were observed in this study where the heating of dobutamine in acidic conditions produced no significant degradation. In contrast, dobutamine is highly susceptible to oxidation in alkaline conditions where the drug is converted to aminochrome at pH 11–13 via a free radical mediated mechanism.17 18 The aminochrome undergoes rapid and complex oxidation and condensation leading to unknown structures, including dark coloured polymers.4 This was observable as a discolouration that was immediately apparent following the addition of 2 M NaOH solution to the dobutamine sample. Dobutamine is also sensitive to photodegradation, possibly via the formation of free radical intermediates similar to the mechanism described for alkaline conditions, leading to red/brown discolouration. Furthermore, as dobutamine is racemic, there is a possibility of differential enantiomer degradation, although this has not been studied to date and is unlikely to be of significance to the findings of this study.
The dobutamine content of the CIVAS syringes declined over the course of the study but generally remained within 5% of the initial concentration. Higher degradation rates occurred at higher temperature and in the presence of light but did not give rise to secondary chromatographic peaks nearing the limit for related substances described in the monograph for ‘Dobutamine intravenous infusion in the Ph. Eur 2010’. In addition to the loss of dobutamine measured by HPLC, a colour change was noted in the syringes. The syringes stored under light were the first to display a colour change, although no additional peaks were observed in the HPLC chromatogram. Discolouration of samples stored at 40°C (protected from light), room temperature (in the presence of light) and room temperature (protected from light) were observed from 35, 28 and 42 days, respectively. The samples stored at 4°C and protected from light showed no discolouration throughout the study period. Interestingly, the colour produced in the absence or presence of light was different (brown versus pink, respectively) and a precipitate occurred in the samples stored protected from light only, perhaps indicating the formation of different products. These changes in appearance limited the acceptability of the CIVAS product under these storage conditions despite dobutamine concentrations remaining above the acceptance limit of <5% change from the initial concentration.11
Conclusion
The stability indicating assay for dobutamine developed in the present study was fit for purpose for the routine QC of CIVAS manufactured dobutamine syringes. Although T95% for dobutamine under this condition was predicted to be 111 days, stability of the product was limited by discolouration despite drug concentrations remaining >95% of label claim. When stored at 4°C protected from light, the dobutamine syringes were stable beyond the nominal shelf life of 28 days allocated by the CIVAS unit. The shelf life for dobutamine syringes stored protected from light at 4°C and room temperature was verified as 42 and 35 days, respectively; this being the period over which measurement was performed to support the kinetically derived stability and the absence of discolouration was confirmed.
Key messages
This work enabled the validation of a cost effective batch production process for dobutamine syringes that possess a sufficient shelf life at ward level, and provided a reliable evidence based measure of shelf life under a variety of storage conditions.
This work enabled the validation of a cost effective batch production process for dobutamine syringes that possess a sufficient shelf life at ward level. While product shelf life may be estimated from published data, this study provided a reliable evidence based measure of shelf life under a variety of storage conditions. Verification of shelf lives contributes to minimising wastage and provides assurance of product quality, thereby enhancing the overall value of the CIVAS to risk management.
Acknowledgments
MT performed these studies during the research project of the MPharm degree at the Department of Pharmacy, King's College London. MT presented this work in part at the British Pharmaceutical Conference and was awarded the best oral presentation by the Joint Pharmaceutical Analysis Group. The authors thank Kevin Weeks at GSTFT for technical support and advice in constructing the manuscript.
Footnotes
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.