Article Text

Abstract
The calcineurin inhibitors (CNIs), ciclosporin and tacrolimus, are the mainstay of immunosuppression in solid organ transplantation. Generic formulations of these drugs are now available. With increasing pressure on healthcare budgets and the consequent need to match health expectations to available resources, substitution with a generic product appears an attractive option to reduce costs. Approval of generic products differs from innovator drugs, and narrow therapeutic index drugs (NTIs; including CNIs) bring their own particular considerations. With NTIs, small variations in drug exposure could result in reduced immunosuppression or drug toxicity with potentially adverse effects on patient outcomes. NTIs are subject to stricter regulatory approval versus many other generic drugs. However, different generic formulations may still not necessarily be therapeutically equivalent in individuals, raising the possibility of significant differences in exposure between products. Although regional recommendations vary, many guidelines emphasise the need for NTI drug substitution to be initiated by the transplant physician, thus ensuring careful therapeutic monitoring and reduced negative patient impact. The need for therapeutic monitoring during generic substitution has important implications for the overall costs of generic treatment as these costs have to be factored in to the potential savings made from using generic formulations. The reduced acquisition costs of generic products may not necessarily translate into lower overall healthcare costs. This article examines the issue of equivalence and interchangeability of NTI drugs used in organ transplantation, the implications of the approval process for generic drugs on treatment efficacy and safety, and the effective management of substitutions between products.
- Transplant Medicine
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The calcineurin inhibitors, ciclosporin and tacrolimus, have been the mainstay of immunosuppressive therapy in solid organ transplantation over the decades since their introduction.1 For these agents, and the more recently introduced mammalian target of rapamycin inhibitors (sirolimus and everolimus), the range of drug exposure between lack of efficacy (under-immunosuppression) and increased toxicity is small, leading them to be described as critical dose drugs or narrow therapeutic index (NTI) drugs.2–4 In addition, these immunosuppressive therapies show a wide degree of variability in pharmacokinetics between individual patients, which means that routine therapeutic drug monitoring is required to ensure optimal immunosuppression.5–8
For many immunosuppressive therapies, generic formulations are now available as an alternative to the original branded product. With increasing pressure on healthcare budgets, and the consequent need to match health expectations to the available resources, substitution with a generic product appears an attractive option to reduce costs. Generics are widely and successfully used across clinical practice; in the Netherlands in 2009, for example, 57% of prescriptions were dispensed as generic products.9 However, NTI immunosuppressive drugs bring their own particular considerations, as small variations in drug concentrations in the blood could result in reduced immunosuppression or increased toxicity, with potentially adverse effects on patient outcomes.2 ,8 ,10
This article builds upon existing knowledge11 by examining the issue of equivalence and interchangeability of NTI drugs used in organ transplantation, the implications of the different approval processes for generic drugs on treatment efficacy and safety, and how substitutions between products can be managed effectively.
Generic drug approval and bioequivalence
The approval process for generic drugs differs from that for innovator products (box 1). While a new drug application (NDA) for an innovator drug requires studies demonstrating safety and efficacy, and postmarketing surveillance data to ensure that any adverse events are tracked effectively, a generic product needs an abbreviated NDA. Provided the generic has the same dosage form, same route of administration and same amount of active ingredient as the innovator, and is produced following good manufacturing practice, the generic product simply has to demonstrate bioequivalence to the branded drug.12 ,13 Safety and efficacy studies are not required as these have already been performed for the original product. Importantly, there is no requirement to demonstrate therapeutic equivalence between the two products; this is assumed based on their bioequivalence.2
The approval process for generic drugs: abbreviated new drug application
Required
-
Same dosage form
-
Same route of administration
-
Same amount of active ingredient(s)
-
Follow good manufacturing practice (GMP) guidelines
-
Bioequivalence
Not required
-
Safety studies
-
Efficacy studies
-
Same excipients
-
The same degree of postmarketing surveillance
-
Demonstration of therapeutic equivalence
Bioequivalence can be regarded as a biological quality assessment test. Both the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines recommend that bioequivalence is measured using a standard single-dose randomised, two-period, two-sequence single-dose design.14 For this reason, bioequivalence studies are typically crossover studies conducted in a small group of usually between 24 and 52 healthy volunteers.15 ,16 In a typical two-by-two or AB/BA design, one half of the group receives the test (generic) product first, followed by a washout period, and then the reference (innovator) drug; for the other half of the group, the order of the products is reversed.17 Although very rarely done, the crossover design can be extended to include more than two treatments per subject in consecutive periods or more than two sequences, that is, so-called, high-order crossover designs (eg, AABB/BBAA or AA/BB/AB/BA).
Blood concentration data are collected for each group, and pharmacokinetic parameters (maximum drug concentration [Cmax], time to reach Cmax [tmax] and area under the concentration/time curve [AUC]) are calculated to determine the extent and rate of exposure. The relative bioavailability of the two formulations is then evaluated from these parameters to determine equivalence derived from standard reference tests (analysis of variance).18 The ratio of the geometric mean value for the test formulation to the mean value for the reference formulation is calculated for Cmax and AUC, and the 90% CI for the mean ratio has to be within a specified interval in order for bioequivalence to be demonstrated.
For most drugs, a difference of 20% between products is used to define bioequivalence,12 although some regulators have introduced stricter guidelines for NTI drugs (as discussed below). Taking values for the reference formulation of 100 and for the test product of 80, the test to reference ratio (80/100) gives the lower limit for the bioequivalence interval of 0.80 or 80%, while the reference to test ratio (100/80) gives the upper limit of 1.25 or 125%. So for bioequivalence to be established, the mean ratio and its 90% CI must lie between 0.80 and 1.25 or 80% to 125%—a difference of ±20%.12
It is, however, important to note that bioequivalence relates to the mean of the data for the study population, so values for individual subjects may lie outside the bioequivalence intervals, even though the mean ratios (and CIs) are within the limits. For example, data from a bioequivalence study comparing generic and branded formulations of ciclosporin showed that almost 40% of individuals receiving the generic formulation were below the 80% limit for the mean ratio for Cmax, with two individuals receiving a maximum drug concentration approximately 40% lower than with the branded formulation (figure 1). Nevertheless, since the 90% CIs of the mean ratios for Cmax and AUC fell within the 80% to 125% limits, the formulation was considered bioequivalent.
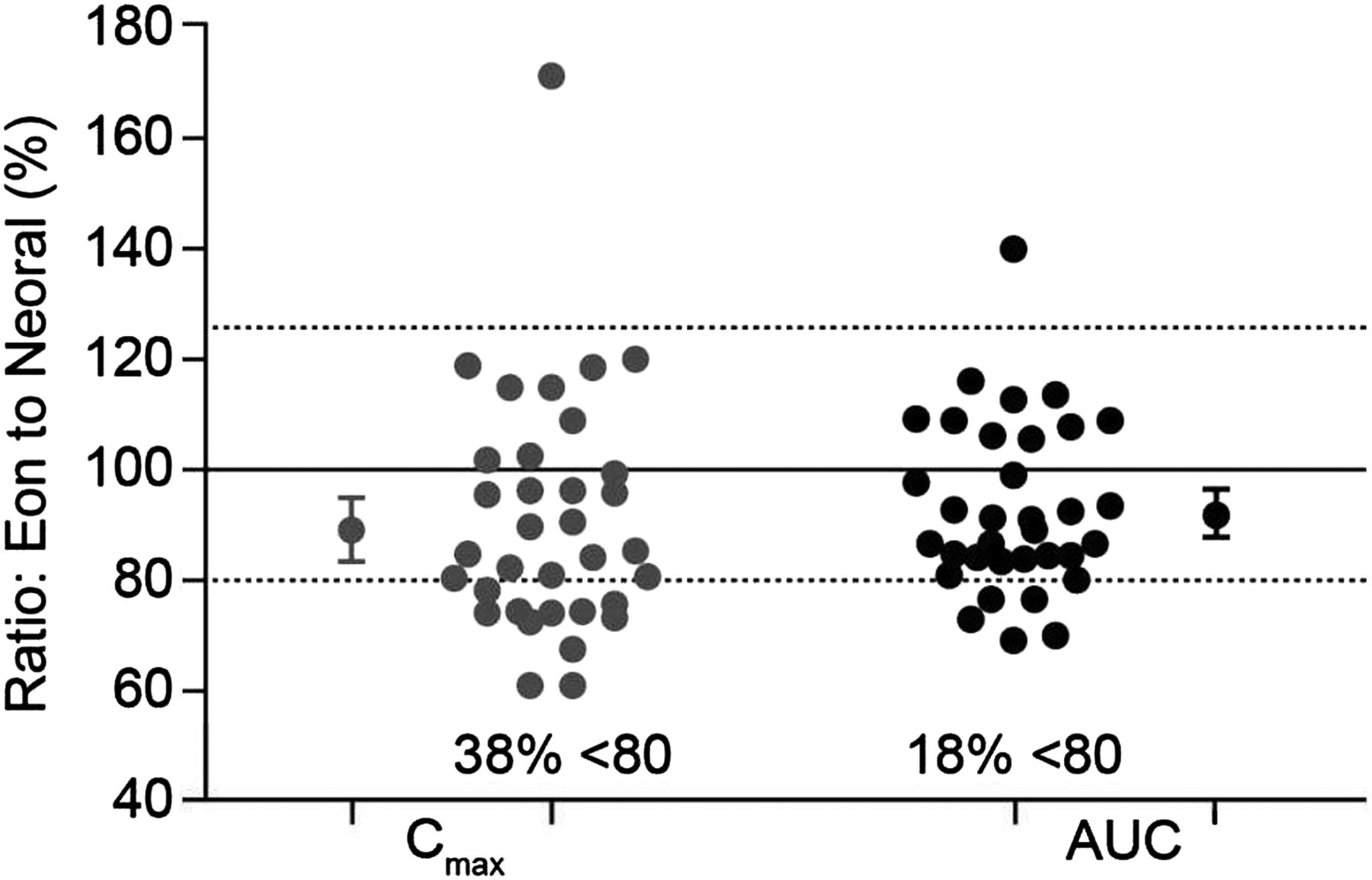
Variation in individual exposure between generic and branded ciclosporin formulations in a bioequivalence study. Data are shown for individual subjects. The large circles show overall mean ratios for Cmax and AUC and encompass the 90% CI of these parameters. AUC, area under the concentration/time curve; Cmax, maximum plasma concentration. Based on study Report Number ANA-97-132, submitted by Eon Labs Manufacturing, Inc, Laurelton, New York (USA) to the Illinois (USA) Department of Public Health (n=34).
Bioequivalence and interchangeability
The approval process for generic drugs also raises the question of interchangeability between different generic formulations as each one only has to demonstrate bioequivalence to the innovator drug. Thus, although two generic formulations (A and B) may each show bioequivalence to the branded product, generic A may not be bioequivalent to generic B, as the 90% CIs for the mean ratios could lie outside the 80%–125% limits (figure 2A). In the included example, a patient who was switched from generic A to generic B would, on average, receive 52% more drug, while conversion from B to A would reduce exposure by 28% on average. Furthermore, these differences could be larger for individual patients. There are, however, a number of mathematical models, which claim to address the potential interchangeability between different generic formulations through indirect comparisons.19 ,20 In one such analysis, Herranz and colleagues (2013) concluded that generic products were not only bioequivalent with the reference product but also with each other; however, the 80%–125% limits used in these analyses are now considered inappropriate for NTI drugs.21
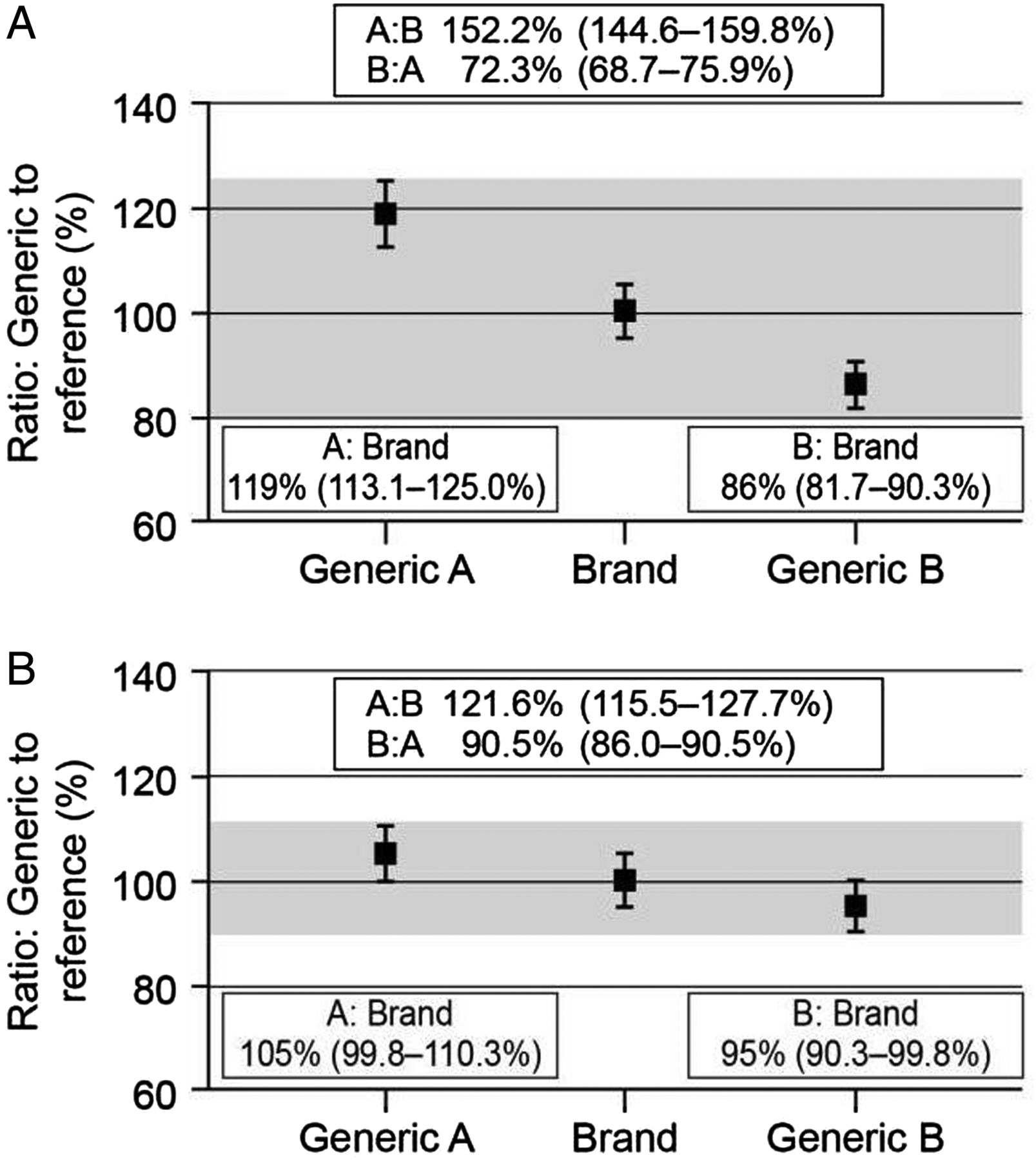
Bioequivalence and interchangeability. (A) Normal bioequivalence limits (80%–125%). (B) Bioequivalence limits for narrow therapeutic index drugs (90%–111%). Generic A and generic B each show bioequivalence to the branded product, but generic A is not bioequivalent to generic B, as the 90% CIs for the mean ratios lie outside the bioequivalence limits.
The potential for such large changes in exposure between different formulations has led some regulatory authorities to introduce tighter bioequivalence limits for NTI drugs or critical dose drugs, such as tacrolimus.3 ,4 ,12 In Europe, for these drugs the limit is ±10%, so the mean ratios for test to reference product (and the 90% CIs) have to be in the range of 90% to 111% to show bioequivalence.3 ,12 Although it is still possible that generic products will not show bioequivalence to one another, the stricter bioequivalence limits mean that potential differences in exposure between the generic formulations are reduced (figure 2B). However, this variability is still a concern, particularly where large numbers of different generic formulations are available for a given drug, as multiple substitutions between the various products could lead to considerable variability in exposure, which may result in poor long-term outcomes.22
Therapeutic equivalence
The approval of generic drugs using bioequivalence studies is based on the fundamental assumption that if two formulations are shown to be bioequivalent, they will provide the same therapeutic effect. In other words, they will be therapeutically equivalent and are therefore interchangeable. However, evidence from studies with ciclosporin, and more recently, tacrolimus, suggests that this is not necessarily the case for the NTI drugs in transplantation.
Initial retrospective analyses of kidney transplant data from the Collaborative Transplant Study database23 suggested that patients treated with generic ciclosporin may have reduced 1-year graft survival compared with those receiving the branded formulation of ciclosporin, Neoral (Novartis Pharmaceuticals).24 Of 17 198 renal transplant recipients who received organs from deceased donors between 1998 and 2000, graft survival was approximately 90% in 16 801 patients who received the branded formulation compared with around 80% in the 397 patients who received the generic drug. Stratification of the data for geographical region in order to reduce bias showed an unfavourable trend for generic ciclosporin in all of the regions assessed, although limited published details on the methodology and observance to treatment prohibit any definitive conclusions to be drawn from this study.23
A retrospective review of 188 de novo kidney transplant patients at a single centre showed a significantly higher rate of biopsy-proven acute rejection (BPAR) with the generic ciclosporin Gengraf (Abbott Laboratories) than with Neoral.24 Patients receiving Gengraf were significantly more likely to have a BPAR episode or a second rejection episode compared with those treated with Neoral (table 1). In addition, significantly more Gengraf-treated patients received an antibody preparation to treat acute rejection than those receiving Neoral (table 1). Although mean 12-h trough concentrations of ciclosporin were similar with the two formulations, patients treated with Gengraf had significantly higher intrapatient variability for ciclosporin trough concentrations than those treated with Neoral (p<0.05).24
Biopsy-proven acute rejection episodes at 6 months post-transplant in adult de novo kidney transplant recipients24
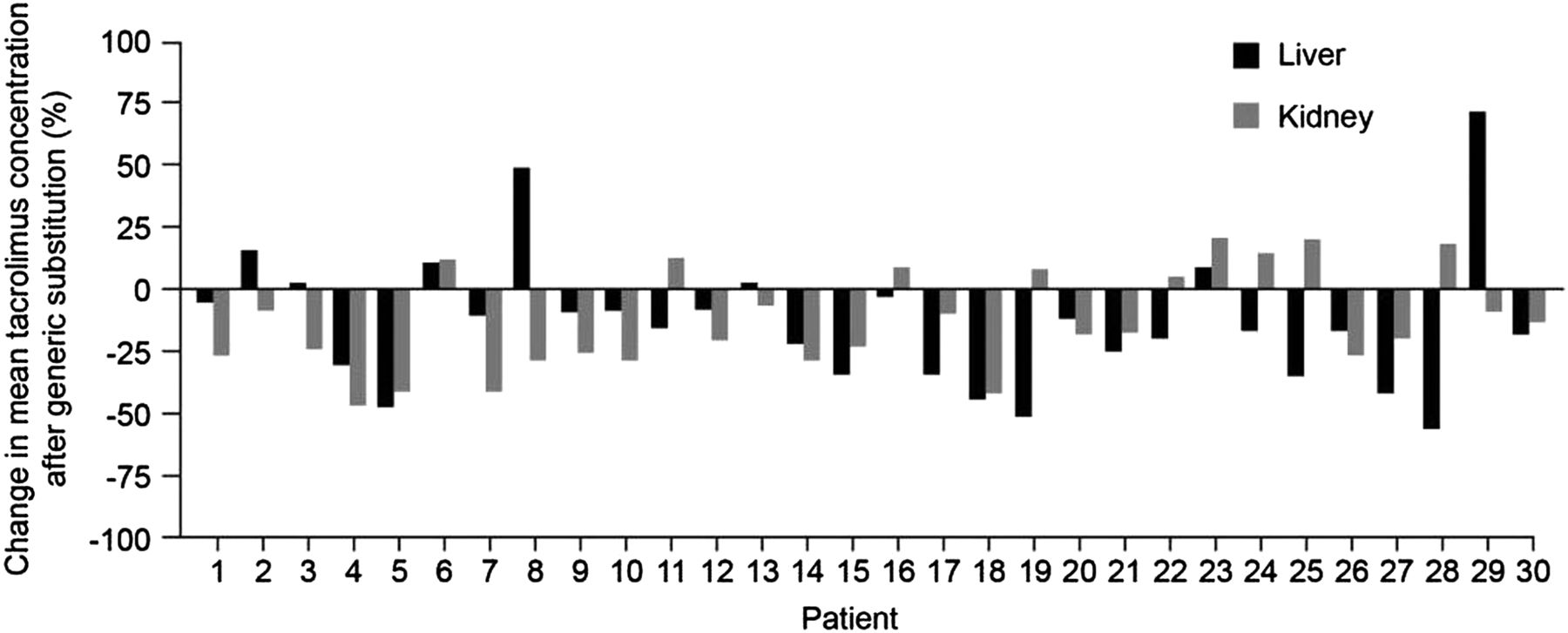
Previous studies have shown that increased within-patient variability in ciclosporin concentrations is associated with poorer outcomes and higher treatment costs following transplantation.25–28 An analysis of 204 transplant recipients showed that the time to chronic rejection over 5 years was significantly longer among patients with less variable ciclosporin concentrations than those with variable concentrations.27 Similar findings have been observed with tacrolimus therapy. A study of 297 kidney transplant recipients at a single centre showed that low within-patient variability in tacrolimus clearance was associated with significantly better graft survival compared with high within-patient variability (figure 3).29 Patients with low variability in tacrolimus trough levels also showed significantly lower acute rejection rates and higher 1-year graft survival than those with high variability in a recent study of over 250 adult kidney transplant recipients.22 Within this setting, other factors, such as interaction with food or other drugs and timing of trough levels, are also important considerations in ensuring low within-patient variability. Data comparing generic and branded formulations of tacrolimus are more limited than for ciclosporin, as generic tacrolimus has only recently become available, whereas generic ciclosporin has been available for over a decade. However, a recent study examined the effects on predose tacrolimus trough concentrations of switching from the branded formulation (PROGRAF; Astellas Pharma) to a generic formulation (Adoport; Sandoz) in 103 clinically stable kidney and liver transplant patients.30 Treatments were switched on a milligram-for-milligram basis, and patients were carefully monitored before and after conversion. Both kidney and liver transplant patients showed marked changes in tacrolimus trough concentrations following the switch to generic tacrolimus. Among the subgroup of 30 kidney and 30 liver transplant recipients who remained on the same dosing regimen throughout the study period, mean reductions in tacrolimus trough concentrations were 0.87 ng/mL and 1.98 ng/mL, respectively, after adjusting for all significant covariates.30 The changes seen in both groups were appreciable, considering the low target trough levels in the study (kidney, 5–7 ng/mL; liver, 6–8 ng/mL). Decreases in tacrolimus blood levels of at least 25% were observed in 40% of the kidney transplant patients and 33% of the liver transplant recipients (figure 4).30 Despite the lower concentration-to-dose (C/D) ratios and a small but significant drop in tacrolimus concentrations, the authors concluded that transplant patients can be safely switched from the branded formulation to the generic tacrolimus formulation ‘provided trough concentrations are closely monitored following the substitution’.30
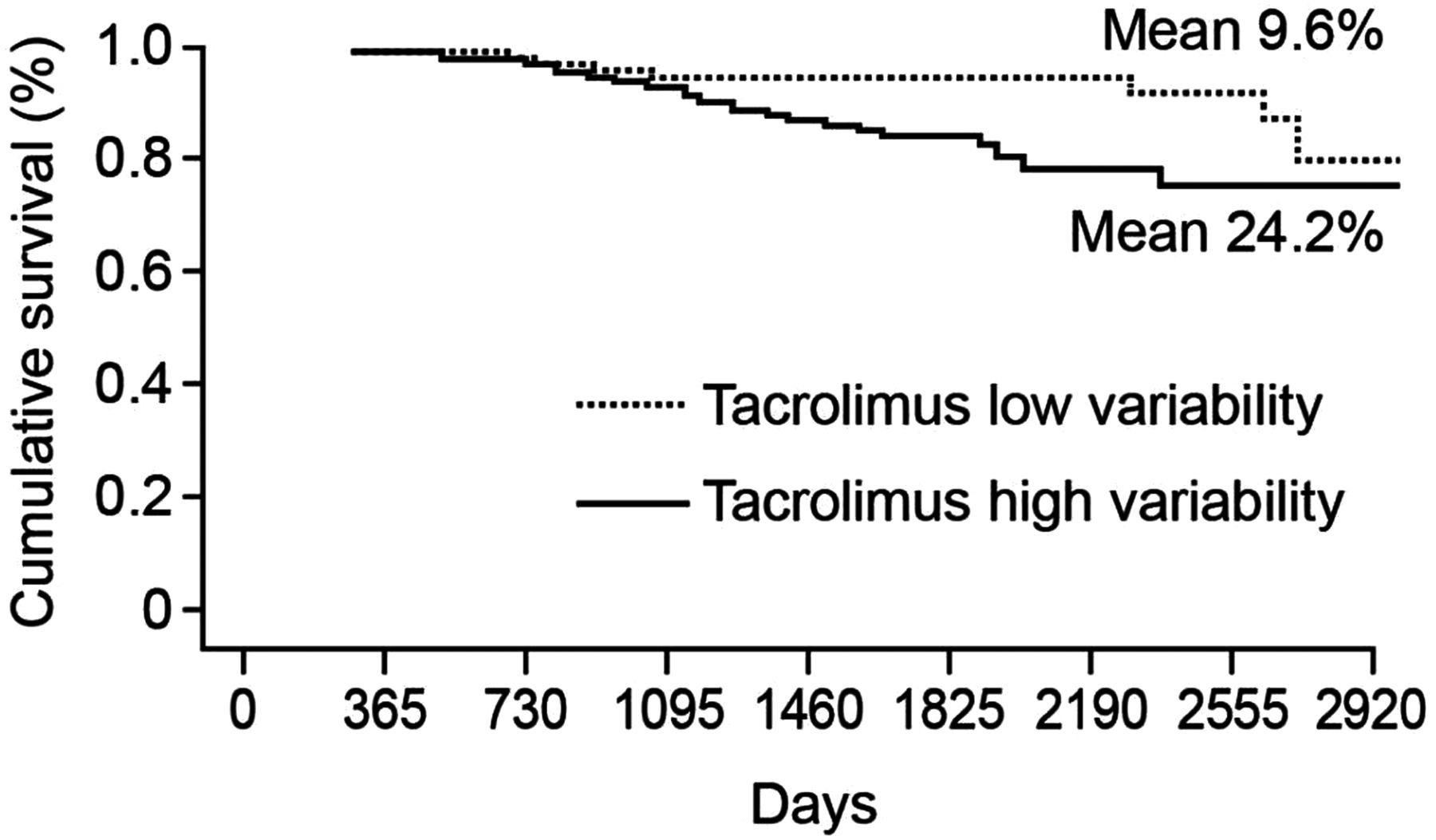
Graft survival for kidney transplant patients with low and high within-patient variability for tacrolimus clearance. Reprinted from Borra et al, 2010.29

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Percentage change in tacrolimus blood trough concentrations following conversion to generic tacrolimus in liver and kidney transplant recipients. Data shown for transplant recipients (kidney, n=30; liver, n=30) whose dosing regimen remained unchanged following 1:1 dose conversion. Adapted from Momper et al, 2011.30
Two subsequent studies31 ,32 have shown that trough levels are similar before and after conversion from branded to generic tacrolimus. However, McDevitt-Potter et al31 reported a substantial increase in the number of dose adjustments required in patients after conversion from the branded formulation to the generic formulation compared with 6 months prior to conversion (21% vs 7%, respectively; p=0.028). Both groups suggest that switching should be performed carefully with vigilance and increased therapeutic drug monitoring, thus recognising that variability between formulations is an issue.31 ,32 With reference to interchangeability between different tacrolimus formulations, the correlation between C12 and AUC across different formulations needs to be established. In the absence of this information it cannot be presumed that when patients are switched between formulations (either branded to generic or from generic to generic formulations) that the same trough levels will achieve the same AUC. Alloway et al33 assessed the correlation between C12 and AUC for conversion from branded formulation to a single generic formulation; this information, however, cannot be applied to other generic formulations. Furthermore, there is a risk for uncontrolled switching between generic formulations where the correlation between C12 and AUC is not established, thereby exposing the patient to substantial risk and inconsistencies in tacrolimus exposure.
These findings are echoed in recent advice from Sandoz to transplant centres in the UK about the use of the generic tacrolimus formulation, Adoport, which states that ‘oral tacrolimus formulations are not interchangeable without careful therapeutic monitoring’ and warns that ‘inadvertent, unintentional or unsupervised switching between different oral formulations of tacrolimus can lead to graft rejection or an increased incidence of undesirable effects’.34 So even though the generic formulation has demonstrated bioequivalence to the branded formulation, the two are not interchangeable without therapeutic drug monitoring.
The need for careful monitoring is also emphasised in an editorial published in response to the study by Momper et al.35 This calls for the introduction of tighter regulations by the US FDA for demonstrating bioequivalence for generic immunosuppressive drugs, together with a requirement for the studies to be performed in transplant recipients (one study for each organ recipient group) rather than healthy volunteers, as drug pharmacokinetics are known to be different in liver and kidney transplant patients. The author concluded that until such changes are in place, patients’ health is being jeopardised if ‘vigilant therapeutic drug monitoring’ is not performed.35 In practice, however, it would be difficult to control for significant confounders, such as the impact of drug interactions on tacrolimus serum concentrations, and for this reason, differences in the drug formulations would be extremely difficult to detect.36
Recommendations for generic substitution
The rise in the availability of generic formulations coupled with concerns over the therapeutic equivalence of NTI drugs has led to a number of transplantation organisations offering guidance on the use of generic immunosuppressive agents.37–39 Typically, these recommendations emphasise the need for the transplant physician to authorise any substitutions.38 ,39 The Kidney Disease: Improving Global Outcomes (KDIGO) clinical practice guidelines advise caution in choosing a generic formulation for treating kidney transplant recipients, as head-to-head comparison data for efficacy and toxicity are generally not available for most generics.37 They state that ‘ideally, a generic formulation should be used only after its safety and efficacy have been established in kidney transplant recipients’. The Efficacy and Safety of PRescribing In Transplantation Group (ESPRIT) website (http://www.esprit.org.uk/) provides information and resources to support the safe use of generic substitution.
The European Society for Organ Transplantation (ESOT) recently published its advisory committee recommendations on the generic substitution of immunosuppressive drugs.39 ESOT makes clear that, as a society, it is not against the use of generic immunosuppressive products. Rather, its aim is to ensure that substitutions occur in a controlled manner, with the appropriate therapeutic monitoring in place to ensure that drug exposure is maintained and outcomes are unaffected. ESOT therefore provides a number of key recommendations regarding generic substitutions, outlined below.
-
Generic formulations that do not meet the current stricter bioequivalence criteria should not be used, while the use of compounds that were approved before the current guidelines were introduced and are still available should be discouraged until they have demonstrated bioequivalence according to the new EMA guidelines.
-
Substitution of the brand name drug for the generic formulation should only be initiated by the transplant physician, to ensure that the appropriate monitoring of drug blood concentrations takes place following the switch, and pharmacists and health insurance providers should refrain from forcing generic substitutions. This aims to avoid the situation where drug exposure is not monitored because the prescriber is unaware of the substitution, leading to the risk of underexposure or overexposure.
-
Repetitive (consecutive) substitutions between different generic formulations of the same drug should be avoided. Prescribers are often unaware of such substitutions, and changes in exposure can be more pronounced than for the change from branded product to generic product (as discussed above). Furthermore, the repeated switching between generic formulations can prove confusing for patients and lead to mistakes with dosing.
-
Patients should be informed about generic substitution and taught how to identify the different formulations of the same drug, and they should alert the transplant physician if an uncontrolled substitution is made. The guidelines emphasise that pharmacists have an active role to play in both informing the patient about the newly prescribed formulation when generic substitution is initiated by the prescribing physician, and protecting the patient from subsequent substitutions.
-
The simultaneous use of different formulations in the same patient should be avoided as this could be confusing for the patient and lead to unpredictable pharmacokinetics and increased variability in exposure.
-
The guidelines also emphasise the role of the transplant community in collecting data on the use of generic immunosuppressive formulations in transplant patients, so enabling a comprehensive picture of all aspects of these drugs (including pharmacokinetics, efficacy and safety) to be assembled.
McDevitt-Potter et al showed that when dose requirements and trough levels are similar between branded and generic tacrolimus, generic substitutions may be associated with cost savings. However, this paper discusses switching from the branded drug to only one generic formulation and the impact of switching to other or between generic formulations was not tested.31 Indeed, there is potential for switching to multiple formulations of generic drugs that enter the market at various time points. The need for careful therapeutic monitoring during generic substitution has important implications for the overall costs of generic treatment, as the costs of monitoring and dealing with patients’ concerns have to be factored in.40 The reduced acquisition costs of generic products may not translate into lower overall healthcare costs if the additional monitoring outweighs the reductions in drug costs. In the absence of data on interchangeability between different generic formulations, it is therefore not possible to conclude on the economic impact of switching from branded to generic formulations.
To date, published direct healthcare cost assessments are limited to generic substitution from product brands. One such US study in de novo renal transplant recipients showed that patients receiving generic ciclosporin incurred significantly higher total healthcare costs over the course of a year compared with those treated with branded ciclosporin, despite initial perceived cost savings associated with the generic formulation.41 Potential savings from reduced drug costs associated with generic substitution therefore need to be evaluated in light of the overall healthcare costs within each healthcare setting.
Conclusions
A large number of generic formulations of immunosuppressive drugs are currently available, with the numbers set to rise over the next few years as more patents expire, while increasing pressures on healthcare budgets are likely to increase the demand for their use. However, approval of generic products differs from innovator drugs. It is based on bioequivalence to the innovator drug (but not to other generic formulations), while therapeutic equivalence is assumed based on bioequivalence. Consequently, different generic formulations may not necessarily be bioequivalent to each other, raising the possibility of significant differences in exposure between different products. In addition, it is not clear how different generic products compare with existing formulations in terms of therapeutic efficacy and the risk of adverse events. Immunosuppressive drugs, such as tacrolimus and ciclosporin, are NTI drugs, and so small changes in exposure could result in increased toxicity or reduced immunosuppression. Conversion to a generic formulation therefore requires careful clinical and therapeutic drug monitoring to ensure that optimal immunosuppression is maintained; hence, generic substitution must only be performed under the close supervision of a transplant specialist. Moreover, the costs of this supervision need to be taken into account when assessing the overall healthcare costs associated with generic substitution.
Acknowledgments
The author would like to thank iS Health for editorial support in the preparation of the manuscript.
References
Footnotes
-
Contributors AJ interpreted the data, was involved in drafting the article, revised it critically once drafted for important intellectual content and approved the final version prior to publication.
-
Funding The editorial services of iS Health, involved in the preparation of this manuscript, were supported by Astellas Pharma Europe Ltd.
-
Competing interests Professor Johnston has been, and is, a consultant for, and owns stock in, several pharmaceutical and biotech companies. He is a director of ESPRIT (Efficacy and Safety of PRescribing In Transplantation)—a not-for-profit company committed to the continued, effective and safe treatment of transplant patients through the education of healthcare professionals and patients. Professor Johnston, his family members and friends use generic and branded drugs. He is also a tax payer and funds, at least in part, the National Health Service in the UK.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial