Article Text

Abstract
Objectives The implementation of dose-banding (DB) in centralised, pharmacy-based cytotoxic drug preparation units allows the preparation of standardised doses in series. The aim of this study was to evaluate the feasibility of DB for the prescribing of ganciclovir (GV) infusion solutions and to investigate the microbiological stability of dose-banded, automatically prepared ready-to-administer GV infusion bags by media-fill simulation tests and sterility tests.
Methods The frequency of prescription of GV doses was retrospectively analysed before and after implementing the DB scheme. Four dose-ranges or ‘bands’ and the corresponding standard doses (250, 300, 350, 400 mg) were identified. The maximum variance was set at ±10% of the individually prescribed dose. The aseptic preparation of a series of GV infusion bags was simulated with double strength tryptic soy broth as growth medium and prefilled 0.9% NaCl polyolefin infusion bags as primary packaging materials. The simulation process was performed with the APOTECAchemo robot on five consecutive days. In total, 50 infusion bags were filled, incubated and stored for 12 weeks at room temperature. The media-filled bags were visually inspected for turbidity after 2, 4, 8, 10 and 12 weeks. Following incubation, growth promotion tests were performed. During the simulation tests, airborne contamination was monitored with settle plates and microbial surface contamination with contact plates. Pooled sterility tests were performed for a series of 10 standard GV infusion bags after a 12-week storage period under refrigeration (2 °C–8 °C).
Results After implementation of the DB scheme, about 60% of the prescribed GV doses were prepared as standard preparations by the robotic system. The number of different GV doses was reduced by 61.8% (76 vs 29). None of the 50 media-filled bags showed turbidity after a storage period of 12 weeks, indicating the absence of microorganisms. The environmental monitoring with settle/contact plates matched the recommended limits set for cleanroom Grade A zones, except in the loading area of the robot. Media fills used for the sterility tests remained clear during the incubation period, thereby revealing sterility. Positive growth promotion tests proved the process’s reliability.
Conclusions A DB scheme for prescribing and preparation of standard GV infusion bags was successfully implemented. Microbiological tests of aseptic preparation of infusion bags in series by the APOTECAchemo robot revealed an adequate level of sterility and a well-controlled aseptic procedure. The sterility was maintained over extended storage periods, thereby encouraging extended beyond-use dating.
- dose-banding
- ganciclovir
- robotic system
- media-fill tests
- microbiological stability
Statistics from Altmetric.com
Introduction
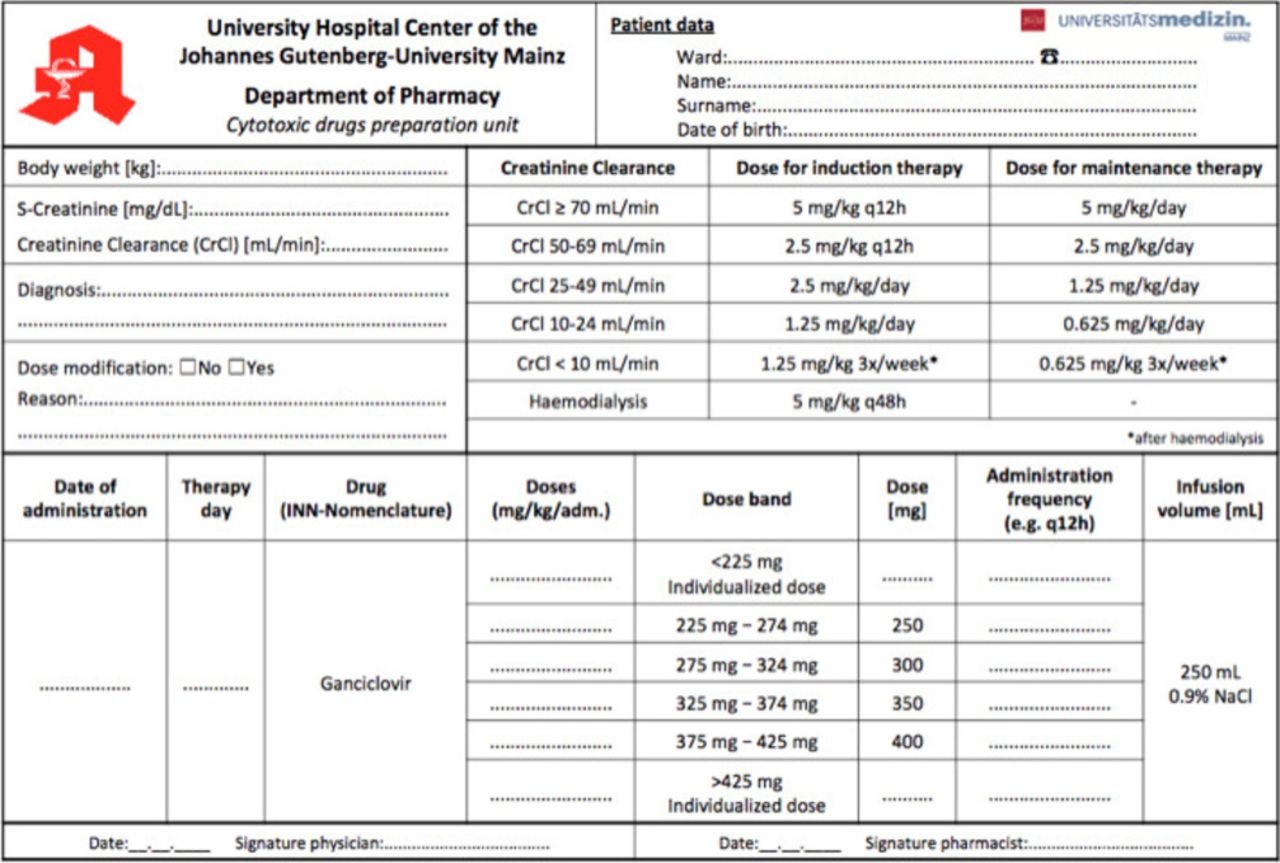
Ganciclovir (GV) is indicated for the treatment of CMV disease in immunocompromised patients and prevention of CMV disease in patients with drug-induced immunosuppression (for example, following organ transplantation or cancer chemotherapy).1 For induction treatment of CMV disease, 5 mg/kg GV are given as an intravenous infusion over 1 hour, every 12 hours for 14–21 days. For maintenance therapy, 5 mg/kg GV may be given once daily for 7 days per week or 6 mg/kg once daily for 5 days per week. In patients with renal impairment, dose modifications according to the creatinine clearance are required.1 A table with the recommended doses for induction and maintenance treatment according to the estimated creatinine clearance (eCrCl)1 is shown in figure 1 as part of our prescribing form.
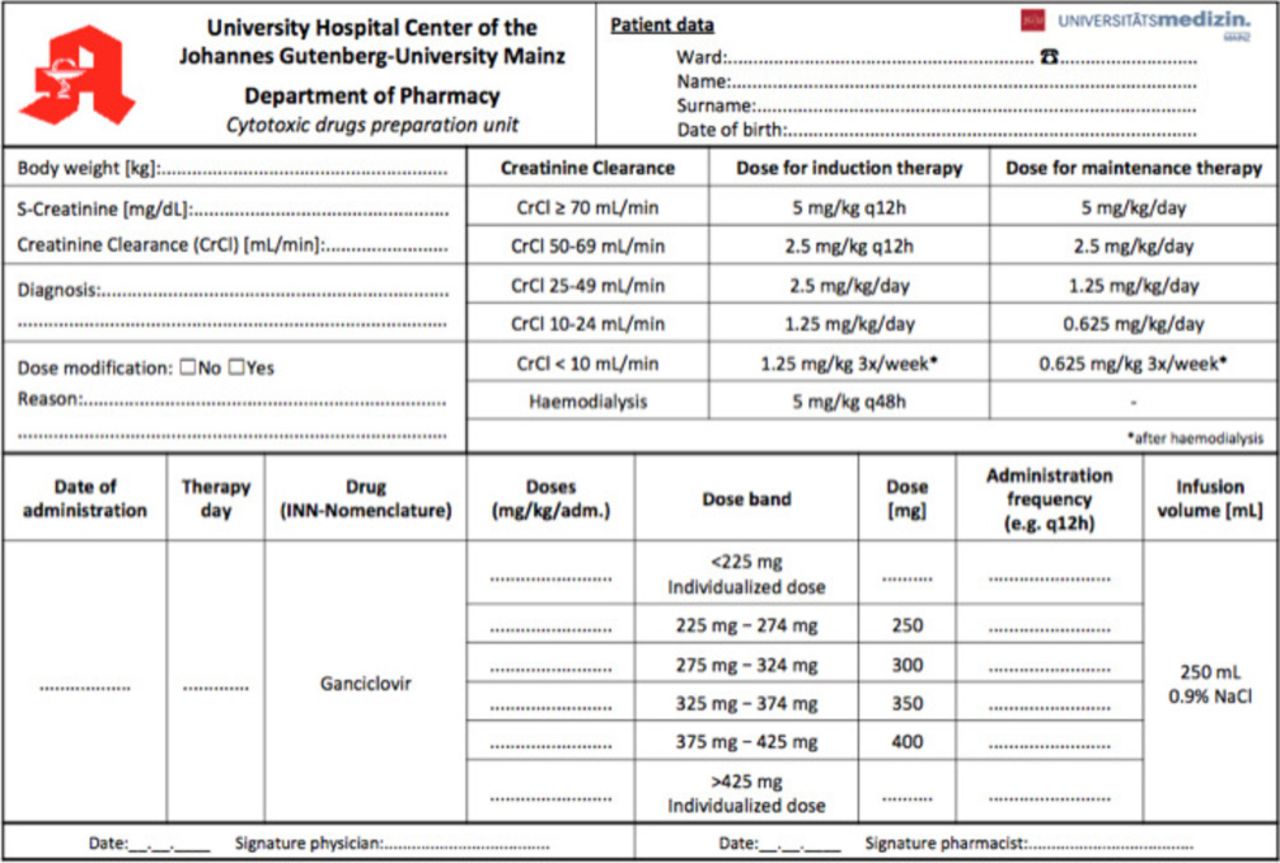
The combined calculation, prescribing and order form for GV preparations after implementation of the dose-banding scheme.
GV is marketed as 500 mg powder for concentrate for solution for infusion that is reconstituted in 10 mL of water for injection to achieve a concentration of 50 mg/mL GV. Prior to intravenous administration, the GV premix solution is to be diluted with 0.9% sodium chloride infusion solution. Since GV is considered a potential teratogen and carcinogen in humans, it should be handled as a hazardous drug.1 2 Moreover, reconstituted GV solutions are alkaline (pH ~11). In order to prevent occupational exposure and to assure sterility of the ready-to-administer infusion solutions, patient individual GV-infusions are often prepared under aseptic conditions in pharmacy-based centralised cytotoxic drug preparation units. Annually about 2000 patient individual GV preparations are produced in the pharmacy department of the University Medical Centre Mainz (Germany). To manage the increasing workload and to optimise the capacity of the preparation unit, dose-banding (DB) of GV infusion solutions was considered. DB is a system whereby individually calculated doses of intravenous cytotoxic drugs are grouped within defined ‘bands’ (dose ranges) and predetermined standard doses (band midpoint) are prepared and dispensed.3 DB allows the preparation of standardised products in series and as stock, efficient automation of the preparation process by robotic systems, and quality control testing prior to administration. Key determinants for dose banding and pre-production of ready-to-use products are extended stability data and a reasonable turnover of the products. Further details regarding the major benefits and drawbacks of DB can be found in the literature.3–8 There are no data about dose banding of GV to be found in the literature except the recently published hint by Guichard et al.9
The aim of this study was to evaluate the feasibility of DB for the prescribing of GV infusion solutions in a single unit form and to investigate the microbiological stability of automatically prepared ready-to-use GV infusion bags by media-fills and sterility tests.
Methods
Retrospective analysis of patient individual GV preparations
Individually dosed GV preparations were retrospectively analysed over an 8-month period (January to August 2015) regarding the potency of prepared doses and the frequency of preparation. The ranges of the most frequently prescribed GV doses were identified. Likewise, after the introduction of the DB scheme, the frequency of prescription of standard and individualised GV doses was analysed over a period of 8 months (September 2015 to April 2016). Data were retrieved from the preparation software Zenzy (Dr Heni Software GmbH, Germany) which was at that time installed in the cytotoxic drug preparation unit of the pharmacy department.
Development of the dose-banded prescribing scheme
According to the ‘target dose’ banding scheme initially four bands and the corresponding standard doses of GV were calculated by the most often prescribed doses and body weight calculations. Each standard dose was defined as the midpoint of the band, resulting in a maximum ±10% variance from the individually prescribed dose (table 1). Prescribing physicians should calculate the patient individual GV dose by the body weight and eCrCl, select the adequate band and thereby the standard dose. An excel sheet was designed as an electronic or pre-printed prescribing and order form (figure1).
Characteristics of the target dose-banding scheme for GV
Aseptic preparation of standardised GV infusion bags in series and quality control
GV standard doses (250 mg, 300 mg, 350 mg, 400 mg) were prepared in prefilled polyolefin infusion bags containing a nominal volume of 250 mL 0.9% NaCl vehicle solution (Freeflex, Fresenius Kabi, Germany). A series of 10 infusion bags per standard dose was aseptically prepared with the robotic system APOTECAchemo (see figure 2, manufactured by Loccioni Group, Italy). Infusion bags were labelled, single packed in plastic bags and heat-sealed. Each series was identified by a ‘lot number’ and stocked under refrigerated storage conditions in the pharmacy department up to 4 weeks.

Picture of the APOTECAchemo (Loccioni Group, Italy) robot and graphical scheme of the locations determined for environmental monitoring with settle plates and contact plates. (L1: loading area; C2: carousel area; W2: balance; W3: surface under the dosing device; W4: gripper of the robotic arm).
According to the standard procedure, sterility tests were carried out by using a rapid microbiological method.10 One aliquot amounting to 2 mL and one amounting to 8 mL was aseptically withdrawn from two randomly collected preparations of a series of 10 and transferred into an aerobic (BD BACTEC Peds Plus Aerobic medium, BD GmbH, Germany) or anaerobic (BD BACTEC Plus Anaerobic medium, BD GmbH, Germany) blood culture bottle. The media were incubated for 14 days at 38 °C in an automated CO2 fluorescent detection system (BD BACTEC 9240 Automated Blood Culture System, BD GmbH, Germany) for microbial growth detection.
Media-fill simulation tests
The aseptic preparation of standardised GV infusion bags was simulated by using sterile tryptic soy broth (TSB) according to the Ph. Eur. 9/2.06.01.00. The preparation of 10 infusion bags in a series was simulated using the robotic system APOTECAchemo. Media fills were performed on five consecutive days (10 preparations per day, Monday to Friday) by a trained operator at the end of the working day.
Purchased vials containing 50 mL pre-prepared double strength TSB (BD Tryptic Soy Broth double strength, Germany, LOT 7178431, expiration date 02/07/2018), prefilled 250 mL 0.9% NaCl polyolefin infusion bags (Freeflex Fresenius Kabi, Germany, LOT 82LC653304, expiration date 02/2020), and 50 mL single-use syringes (Drug Compounding Dosing Device ASN-50, Loccioni Humancare, Italy, LOT 170407, expiration date 03/2022) were loaded in the carousel of APOTECAchemo. After automated withdrawal of the superfluous vehicle solution from the infusion bags, in total 125 mL of TSB were withdrawn from the 50 mL vials and injected into the infusion bag using a 50 mL syringe. The final volume in the infusion bag amounted to 250 mL (125 mL TSB +125 mL vehicle solution). In total, 50 media-fill products were prepared by using 125 vials nominally filled with 50 mL TSB and 50 infusion bags.
Before starting the preparation process, the outer surface of each vial was disinfected by immersion in a disinfectant solution (Terralin, Schülke&Mayr GmbH, Germany). The surface of the rubber stoppers of each vial, as well as the injection port of each infusion bag were wiped with Perform advanced Alcohol EP (Schülke&Mayr GmbH, Germany) in the loading area of APOTECAchemo and inserted in the carousel. After preparation and unloading, the media-fill products were labelled, single packed into plastic bags, heat-sealed and incubated.
Incubation and analysis – The media-fill products were incubated over 2 weeks according to Ph. Eur. 9/2.06.01.00 and stored for 12 weeks (maximum intended storage interval) at room temperature (20°C–25°C). In order to suspend microorganisms adhering at surfaces and ensure contact with all surfaces, the media-fill products were gently mixed twice per week during the storage period. All samples were visually inspected for turbidity by the unaided eye after 2, 4, 8, 10 and 12 weeks.
Growth promotion test – The purchased TSB vials were accompanied by a certificate, which documented that growth promotion tests were accomplished for the batch. In addition, after 4 weeks of incubation, growth promotion tests of the media fills were performed. Two samples per production day were randomly collected and, overall, 10 media-fill products were inoculated with S. epidermidis KH6 (obtained from a patient sample) or S. aureus ATCC6538 suspensions. Each bacteria strain was suspended in PSB (phosphate buffer solution), adjusted to 103 colony-forming units (CFU)/mL, and used to inoculate five samples. The inoculation was performed by adding 2.5 mL of bacteria suspension to each infusion bag filled with 250 mL growth medium in order to achieve a final concentration of 10 CFU/mL, i.e. 2500 CFU/infusion bag. The inoculated samples were incubated for 2 weeks at room temperature and visually inspected for turbidity every day. Preparation of the bacteria suspensions and inoculation were carried out at the Institute of Medical Microbiology and Hygiene, University Medical Centre Mainz (Germany).
Environmental monitoring
During the media fill simulation tests, airborne contamination was monitored by positioning settle plates (CASO-Agar, heipha Dr Müller GmbH, Germany, LOT880701, expiration date 13/01/2018) at three predefined locations of the APOTECAchemo, see figure 2), that is, loading area (L1), carousel area (C2) and working area (W3) over a 4-hour interval, each.
At the end of each production day, microbial surface contamination was tested with contact plates (CASO-Abklatschagar Tryptic Soy Contact Agar, heipha Dr Müller GmbH, Germany, LOT 919506, expiration date 23/02/2018). The sampling locations corresponded to the surface of the loading area (L1), surface of the balance (W2), single-use mat under the dosing device (W3) and the gripper of the robotic arm (W4) (see figure 2).
In addition, fingerprints were performed from gloved fingertips and thumbs of both hands by pressing on CASO-Contact plates Tryptic Soy Contact Agar tryptic soy (details given above) for at least 5 s at the end of a 20 min glove-wearing period. Fingerprints were collected once during each media-fill test.
Sterility test of standard GV infusion bags
A series of 10 GV infusions bags containing 250 mg of GV (Ganciclovir HEXAL, Hexal AG, Holzkirchen, Germany, LOT GK7722, expiration date 31/08/2019; Freeflex, Fresenius Kabi, Germany, LOT 82KM653305, expiration date 30/11/2019) was aseptically prepared and stored under refrigeration (2 °C–8 °C) for 12 weeks. After the storage period, sterility tests were performed according to the Ph. Eur. 9/2.06.01.00 by a certified laboratory (Labour L+S AG, Bad Bocklet, Germany). From each of the 10 GV-infusion bags, 50 mL were filtered via mixed cellulose esters (MCE) membranes (nominal pore size of 0.45 µm) using SteritestTM EZ device (Merck KGaA, Germany). Filter membranes were incubated with soya-bean casein digest broth (SCD) for aerobic bacteria and fungi and fluid thioglycollate medium (FTM) for aerobic, micro-aerophilic, and anaerobic bacteria. SCD and FTM were incubated for 14 days at 20°C–25°C and 30°C–35°C respectively. The media were examined for macroscopic evidence of microbial growth at regular intervals and at the end of the incubation period.
Growth promotion tests were performed by inoculation of the media with challenge microorganisms. The microorganisms used were C. sporogenes and B. subtilis for FTM and SCD respectively. After inoculation, the media got incubated for 5 days at room temperature and visually inspected for turbidity every day.
Results
Evaluation of the dose-banding scheme
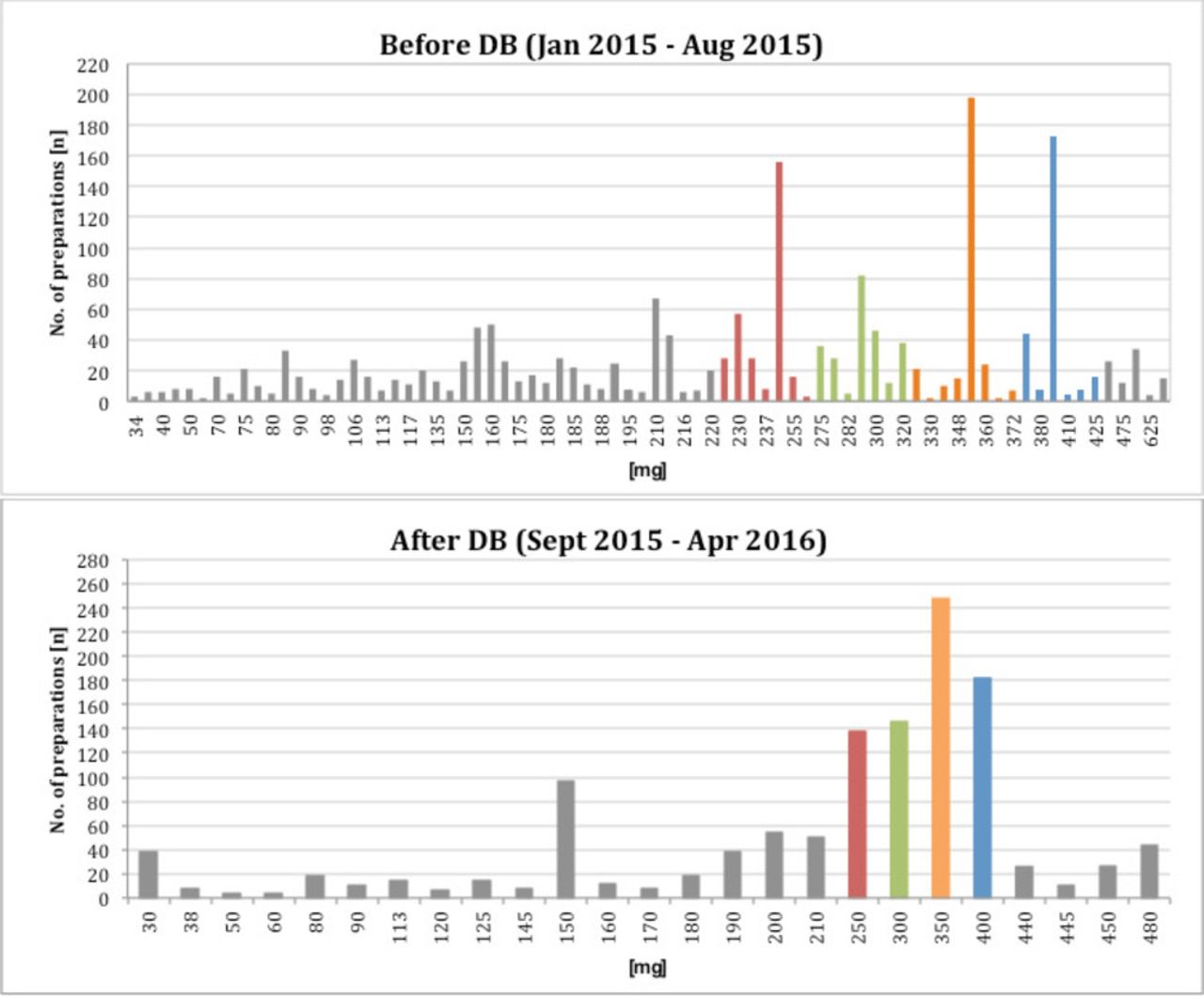
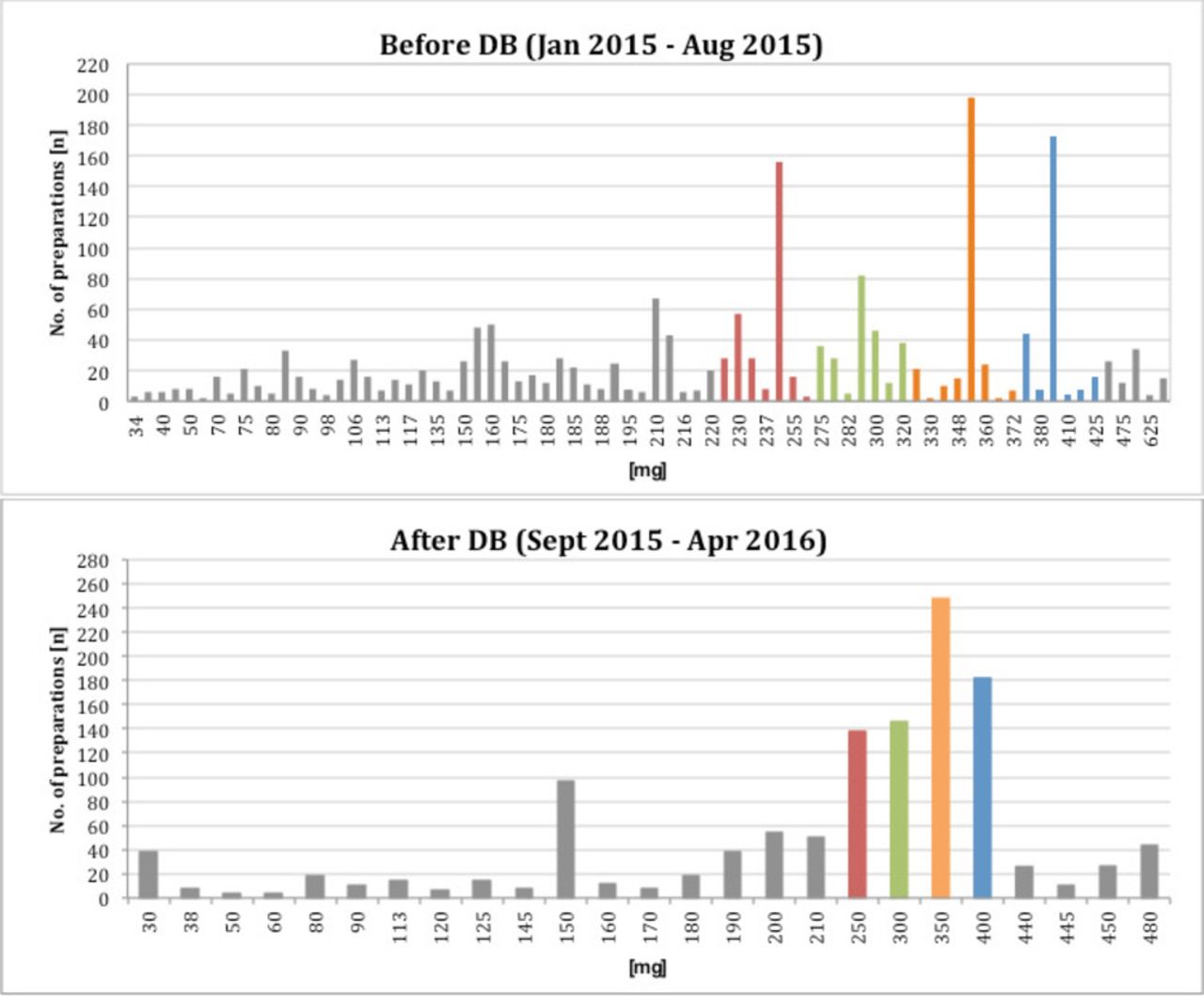
Before implementing the DB scheme, a total of 1886 ready-to-use GV infusion bags corresponding to 76 different doses were prepared during an 8 months period. During the 8 months period following the implementation of the DB scheme, about 60% of the 1225 prescribed doses were covered by the of four standardised preparations. The number of different doses was reduced from 76 to 29 (25 individual and four standard doses). Patient specific doses had to be prepared for patients with body weight >85 kg (single dose >425 mg GV) and patients suffering from renal insufficiency or children (single dose <225 mg GV) (see figure 3). Based on these calculations and agreement of the prescribing physicians a specific calculation, prescribing and order form for GV therapies was implemented. The form is shown in figure 1. It can be used electronically or filled manually in a printed version.
{kind=link}
{kind=link}
{kind=link}
Potency of prepared doses and frequency of preparation before and after implementing the DB scheme.
Media-fill simulation tests
None of the 50 media-fills simulating the automated preparation of standardised GV infusion bags showed turbidity after the incubation period of 2 weeks and up to a storage period of 12 weeks indicating the absence of microorganisms. All products remained clear and light-amber solutions without colour-change. Of note, the growth promotion tests of the media fills performed after 4 weeks of storage, indicated reliability. The inoculated strains (S. epidermidis, S. aureus) caused turbidity in all samples already after 5 days of incubation and till to the end of the 2 weeks observation period (see table 2).
Results of the media-fill simulation tests during automated preparation in APOTECAchemo and visual inspection after 2, 4, 8, 10, and 12 weeks of incubation
Environmental monitoring
The results of the environmental monitoring test performed in parallel to the media-fill simulation tests are presented in table 3. In the carousel area and the working area of the APOTECAchemo robot, the number of CFUs counted on settle/contact plates were below the limits set for EU-GMP Grade A zones (<1 CFU/plate). However, in the loading area, on average four CFUs were detected on settle plates and 12 CFUs on surface contact plates. The very high numbers of CFUs on settle plates and contact plates observed on day 1 of testing can be categorised as outliers. Otherwise the number of CFUs observed on day 2 to 5 in the loading area are acceptable.
Airborne and surface contamination during automated media-fill simulation tests in the APOTECAchemo over 5 days
Sterility test
Pooled sterility tests of 10 GV infusion bags prepared in the APOTECAchemo revealed sterility. Aerobic and anaerobic media remained clear during the incubation period. The microorganisms selected for the growth promotion tests (C. sporogenes and B. subtilis) caused turbidity after 2 days of incubation.
Discussion
Implementation of the dose-banding scheme
DB of cytotoxic preparations is not yet very common in Germany. The reason is that in the past most antineoplastic drugs were investigated in clinical trials in patient individual doses calculated by the body surface area (BSA). Although, the evidence of BSA-related dosing is meanwhile questioned, the dosing method is still carried out in accordance with the clinical trials. Thereby the workload of centralised cytotoxic preparation units in pharmacies is increasing and potentially the risk of compounding errors.11 To optimise the capacity planning of our cytotoxic preparation unit, we considered dose DB of the antiviral drugs and chose GV to start with. GV qualifies for DB by the high number of preparations, the long duration of treatment (at least 2 weeks) with twice and once daily dosing, and proven long-term physicochemical stability of the ready-to-administer infusion bags.9 12 13
The target DB scheme was chosen for the calculation of standard doses and the bands. In accordance with the retrospective analysis of nearly 2,000 BSA-based patient individual preparations, four standardised infusion bags were defined. The aim was to develop single unit doses with acceptable variance between the calculated patient individual dose and the standard dose. Additional manipulations on the ward to achieve the banded dose or combination of multiple infusions should be avoided. For antineoplastic drugs in general, a variance of 5% and for monoclonal antibodies the variance of 10% between the prescribed dose and the banded dose is accepted clinical practice in the UK.4 In our case, the dose variance amounted to minimal 6.2% and maximum 10% for the different bands. The proposed dose bands were agreed by the medical staff, because of the broader therapeutic window of GV compared with antineoplastic drugs and the approved dose variance of 20% (5 mg/kg or 6 mg/kg) during maintenance treatment.1 The retrospective analysis after the introduction of DB showed that the number of GV doses <225 mg was increasing. Therefore, another band (190–224 mg) and the corresponding standard GV infusion bag with 210 mg was established.
DB enables the preparation of standard infusion bags in series and as stock. Quality control testing, such as quantitative analysis of GV and sterility tests can be done prior to administration.14 In our setting the automated preparation of the GV standard doses is more accurate and less error-prone than manual preparation.15–19 Dosing accuracy of the robotic system is verified by gravimetric control of the measured volume and automated documentation of the deviation rate from the target value. The deviation limits were set at ±5% for the GV preparations. Barcode recognition and photographic recognition of the source products ensure the identity of the brand products used.20 Since in-process controls and process documentation are given, parametric release is acceptable.
Reconstituted and diluted GV infusion solutions are known to be physico-chemically stable over prolonged periods. Parasrampuria et al 12 showed that GV admixtures (0.9% NaCl, 5% dextrose vehicle solution) of the concentration 1 and 5 mg/mL are stable for at least 35 days at room temperature or under refrigeration. Phaypradith et al 13 reported stability of GV for at least 1 year at −20°C, 80 days at 4°C and 1 week at room temperature (concentration from 0.004 to 7 mg/mL, 0.9% NaCl vehicle solution, polypropylene syringes or PVC infusion bags). In a recent study, the stability of GV infusions (concentration from 0.25 to 5 mg/mL, 0.9% NaCl vehicle solution) was shown to be at least 185 days stored in polypropylene infusion bags or syringes either at room temperature or under refrigeration.9 Initially we planned a 12-week storage period under refrigeration. Referring to the recently published stability data we switched to room temperature storage because of limited refrigerator capacity and more convenient handling.
However, when the beyond-use dates of ready-to-use preparations are defined, microbiological stability is also to be taken into account.21 22 Sterility and the absence of microorganisms depend on the quality of aseptic procedure and the growth supporting characteristics of the products. In previous growth promotion tests we were able to show, that microorganisms could subsist in solutions of cytotoxic and non-cytotoxic drugs, and proliferate when transferred to proper growth media.23–25 However, during these tests the GV samples showed moderate bacteriostatic and antifungal activity.25 There is no preservative contained in the GV formulation, but the alkaline pH value might contribute to the growth inhibition of microorganisms.
According to the guidelines for Good Aseptic Preparation Practice in pharmacy departments,10 26 the validation of the aseptic procedures should be done by simulating the preparation process with nutrient media (media fills) and environmental monitoring. Extensive tests by simulating the filling of 50 mL syringes with media in the APOTECAchemo robot revealed sterility of the test products.27 In addition, media-fill tests are performed on a regular basis demonstrating the adequacy of the existing aseptic conditions and procedures. Additional media-fill tests were performed to simulate the preparation of GV standard infusions in series by the robot APOTECAchemo. The preparation process of standardised products in pre-filled infusion bags was imitated as closely as possible. All critical steps were included, except the reconstitution of the powder. Simultaneous environmental monitoring revealed in elevated numbers of CFUs in the loading area of the robot and on the fingerprints of the operator on Day 1. This was most probably caused by using media vials bearing a high bioburden on the outer surface of the phials. We made this observation already in the past and requested intensive cleaning and disinfection of the vials. From time to time we recognise such outliers. These observations emphasise the importance of intensive cleaning and disinfection after performing media-fill tests.
The results of supplemental sterility tests of GV standard infusions confirmed the absence of viable microorganisms in the final products. As the tests were performed after 12 weeks' storage, these findings confirm that the storage conditions (2 °C–8°C) and the secondary packaging (heat-sealed plastic bags) ensure maintained sterility of the final products over extended storage periods.
Sterility of the GV standard infusion bags is routinely checked by rapid microbiological methods. Tests are performed for each series of GV products as described above in the Methods section. Results of routine simulation tests and ongoing environmental monitoring by microbiological methods are considered, when a GV series is released.
Overall, the study proved feasibility and a positive impact of DB for GV preparations in clinical practice. After the introduction of DB, the number of patient individual doses decreased drastically. Meanwhile we stock standard GV infusion bags on the wards. By this measure CMV treatment can be started immediately at any time. Furthermore, the DB scheme and preparation method was applied to foscarnet preparations.
Conclusions
A target DB scheme for prescribing and centralised preparation of standard GV infusion bags and patient-specific doses at the extremes of the dose-band range has been successfully implemented. Feasibility and practicality of a specific excel-based form for the calculation, dose rounding and ordering of GV infusions were given. With four and, late,r five standard infusion solutions to be administered as single unit doses, acceptable variances of the dose bands were achieved. Microbiological tests of automatically in series prepared GV infusion bags revealed an adequate sterility level and a well-controlled aseptic procedure. The sterility was maintained over extended incubation and storage periods, thereby encouraging extended beyond-use dating up to 12 weeks. The same DB concept is applicable to other antiviral drugs, for example, foscarnet.
What this paper adds
What is already known on this subject
The implementation of a DB scheme and dose standardisation facilitate the optimisation and the capacity of pharmacy-based, centralised cytotoxic drugs preparation units, especially those with considerable daily workload.
Ganciclovir infusion solutions are known to be physico-chemically stable over prolonged periods.
Automated preparation with robotic systems ensure adequate sterility level, a well-controlled aseptic procedure, automated in-process controls and automated process documentation.
What this study adds
A feasible and practicable DB scheme for prescribing and preparing the antiviral GV.
Automated aseptic preparation of infusion bags in series by using a robotic system.
Extended beyond-usage date (up to 12 weeks) of automatically prepared infusion bags containing GV.
The method analysed can be applicable to other antiviral drugs for parenteral administration.
Acknowledgments
We are grateful to the Institute of Medical Microbiology and Hygiene, University Medical Centre Mainz and the technicians of our Pharmacy Departments for technical support.
References
Footnotes
EAHP Statement 3: Production and Compounding.
Contributors IK and MF carried out the experiment and wrote the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.