Article Text

Abstract
Objectives To design and execute a comprehensive microbiological validation protocol to assess a brand-new sterile compounding robot in a hospital pharmacy environment, according to ISO and EU GMP standards.
Methods Qualification of the Class-A inner environment of the robot was performed through microbial air and surface quality assessment utilising contact plates, swabs and particulate matter monitoring. To evaluate the effectiveness of the microbial decontamination process (UV rays) challenge test against Pseudomonas aeruginosa, Staphylococcus aureus, Bacillus subtilis spores and Candida albicans was used. The challenge Media Fill test was used to validate the aseptic processing.
Results After 3 hours, no microorganisms retained viability. Monitoring inside the equipment evidenced complete absence of microorganisms. The Media Fill test was always negative.
Conclusions According to our results, the APOTECAunit meets the requirements for advanced aseptic processing in the hospital pharmacies and the pharmaceutical industry in general, providing advantages in terms of safety for patients compared with conventional procedures of parenteral preparation production. The protocol has demonstrated to be a comprehensive and valuable tool in validating, from a microbial point of view, a sterile-compounding technology. This study might represent an important benchmark in developing a contamination control strategy, as required, for example, in the Performance Qualification of the GMP in the case of drug manufacturing.
- drug manufacturing / preparation / compounding
- aseptic preparation
- compounding robots
- sterile production
- dispensing robots
- good distribution practice (gdp)
Statistics from Altmetric.com
- drug manufacturing / preparation / compounding
- aseptic preparation
- compounding robots
- sterile production
- dispensing robots
- good distribution practice (gdp)
Introduction
The International Pharmacopoeia requires that parenteral preparations shall fulfil requirements for sterility, pyrogens, particulate matter and other contaminants, since these preparations are intended for direct administration into the blood vessel, organs or tissues.1
The implementation of robotic devices for the intravenous (IV) medication compounding has been proposed to be powerful tools in accomplishing the aseptic processing due to the high repeatability of operations and movements that avoids contacts between critical components and ensures error prevention.2
Nevertheless, although the presence of IV compounding robots in hospitals has led to a significant revolution in clinical practice, their use has mainly occurred in the preparation of IV anticancer drugs as a result of high toxicity and low therapeutic index of certain chemotherapeutic drugs.3–7 Indeed, manual compounding is still the most common practice for the preparation of non-hazardous (n-Hz) IV medications such as antibiotics, anti-emetics, diuretics, cortisones and so on.8 This latter process, performed by either technicians in the hospital pharmacy or by nurses in the hospital wards, can lead to undetectable medication errors and microbiological contamination.9–12
In 2015 the University Hospital of Ancona initiated a project to centralise and automate the compounding of n-Hz medications. As a first step, five hospital departments were selected to investigate the prescribing habits and all medical records of patients admitted in June 2014 to those five pilot departments were retrospectively examined. Particular attention was focused on class of drug, active ingredient, related dosage, dilution, posology and chemical stability of the prescribed IV medications.13 This preliminary study demonstrated the technical feasibility of the project and generated an initial list of the most suitable drugs for study. In April 2016, the first prototype of an IV-compounding robot was installed in a dedicated positive pressure Class-C cleanroom, followed by an intensive 2-month validation in a clinical setting. The results of these studies are reported herein.
This study reports the microbiological validation process of an IV compounding robot, APOTECAunit, for the preparation of sterile n-Hz medications, according to ISO 14 644–1, ISO 13408 and EU GMP standards. It represents the Performance Qualification required by Good Manufacturing Practice (GMP) to approve the robot for patient use in hospital.14 In a previous study, we assessed the efficacy of ultraviolet (UV) irradiation to maintain sterility conditions of another IV-compounding robot, whereas this current study includes three additional aspects: microbial qualification of the Class-A inner environment of the robot; the effectiveness of the automatic microbial decontamination process of the system; and the aseptic processing validation through the challenge Media Fill test.14 15
UV irradiation is a common sanitising agent to inactivate microorganisms in compounding robots. However, the effectiveness of UV disinfection depends on a number of factors such as the power of the UV source, exposure time, distance of microorganisms from the source, microorganism resistance, and presence of particulate and shadow zones 16 17
The Media Fill process simulation is a standard method of assessing and validating the aseptic technique of the pharmaceutical manufacturing, through a challenge test methodology. For sterile solutions, process simulation is conducted using microbiological growth media in lieu of products as the principal method available to ensure that the aseptic process is functioning as intended. The process simulation is carried out under conditions that simulate routine manufacturing procedures and includes all the production steps.
This protocol provides useful information to better understand and design a microbial contamination control strategy for a sterile technology assessment in a clinical environment.
Material and methods
Compounding robot
The IV robotic system used in the University Hospital of Ancona for the sterile compounding of n-Hz drugs is APOTECAunit (figure 1). It is designed for high productivity of both batch production and patient-specific preparations. The equipment has two main areas: the loading area that houses a capacious automatic warehouse; and the compounding area, where a robotic anthropomorphic arm admixes drugs into a closed and microbiologically controlled area, with a vertical laminar airflow and an internal positive pressure, without any human intervention. The incoming/outgoing materials are checked and registered with sensors, photocells, vision system, high precision scale and barcode readers. The automatic barcode labelling of the final products guarantees the traceability along the whole process.

The APOTECAunit robotic system. 1. Loading area, 2. Internal storage area, 3. Compounding area, 4. Final products unloading area, 5. Air-treatment system with HEPA filters.
The system is equipped with seven UV lamps distributed in the internal chambers to ensure the decontamination process. The characteristics of the UV lamps are: peak emission 254 nm; irradiance at 1 m 22 mW/cm2; and radiant flux 2.4 W. The UV irradiation occurs at the end of the production activity, with the system empty and isolated from the external environment.
Qualification of the Class-A inner environment of the robot
A comprehensive environmental monitoring programme was performed to assess the expected EU-GMP Class-A of the inner areas of the robot.14 The qualification consists of a microbial quality assessment of air and surfaces, as well as airborne non-viable particle monitoring, according to ISO14644-1 and EU GMP Annex 1.
Viable particulate air sampling was carried out utilising settle 90 mm Petri dishes. Two series of plates with Tryptic Soy Agar (TSA) were set up for bacteria and fungi, and located in 10 different positions as shown in figure 2A. The monitoring was performed ‘in operation’ during each run of the Media Fill production cycle. The plates were incubated at 36°C for 48 hours and at 25°C for 7 days for bacteria and fungi, respectively, and examined for growth every day, according to PIC/S Recommendation.18 Although active air sampling is preferred, settle plates were selected due to the difficulty of placing active air samplers without interfering with the operation.

Environmental monitoring. (A) Settle plates in the loading area; (B) Swab sampling; (C) Contact plate sampling; (D) Particle counter probe inside the compounding area.
Surface sampling was performed ‘at rest’, using swabs and contact plates depending on the surface, immediately before and after each run of three production cycles. Two series of moistened swabs were streaked in nine different positions inside the equipment (figure 2B). Each swab was then inoculated onto plates containing TSA and incubated as described for settle plates. The contact plates filled with TSA were pressed on eight flat surfaces inside the equipment as shown in figure 2C and then incubated.
Non-viable particle monitoring was executed ‘at rest’, before and after each run, and ‘in operation’ during the whole production cycle described in the ‘aseptic processing validation’ paragraph (figure 2D). The particle counter used was a Lighthouse Solair 3200, with sampling volume of 50 L/min a laser diode as source and a sensitivity down to a particle size of 0.3 µm. At first, the zero-count level was fixed using the purge filter (<1 count/5 min.). The particle sampling (from 0.3 µm to 10 µm diameter) was carried out for 1 min and repeated three times using an isokinetic sample probe positioned in six selected locations (figure 3). The selected location for the ‘in operation’ monitoring was that on the left of the dosing device because it was closer to the most critical point. The sampling was extended to 3 min (average time for a single preparation) and repeated in continuous.
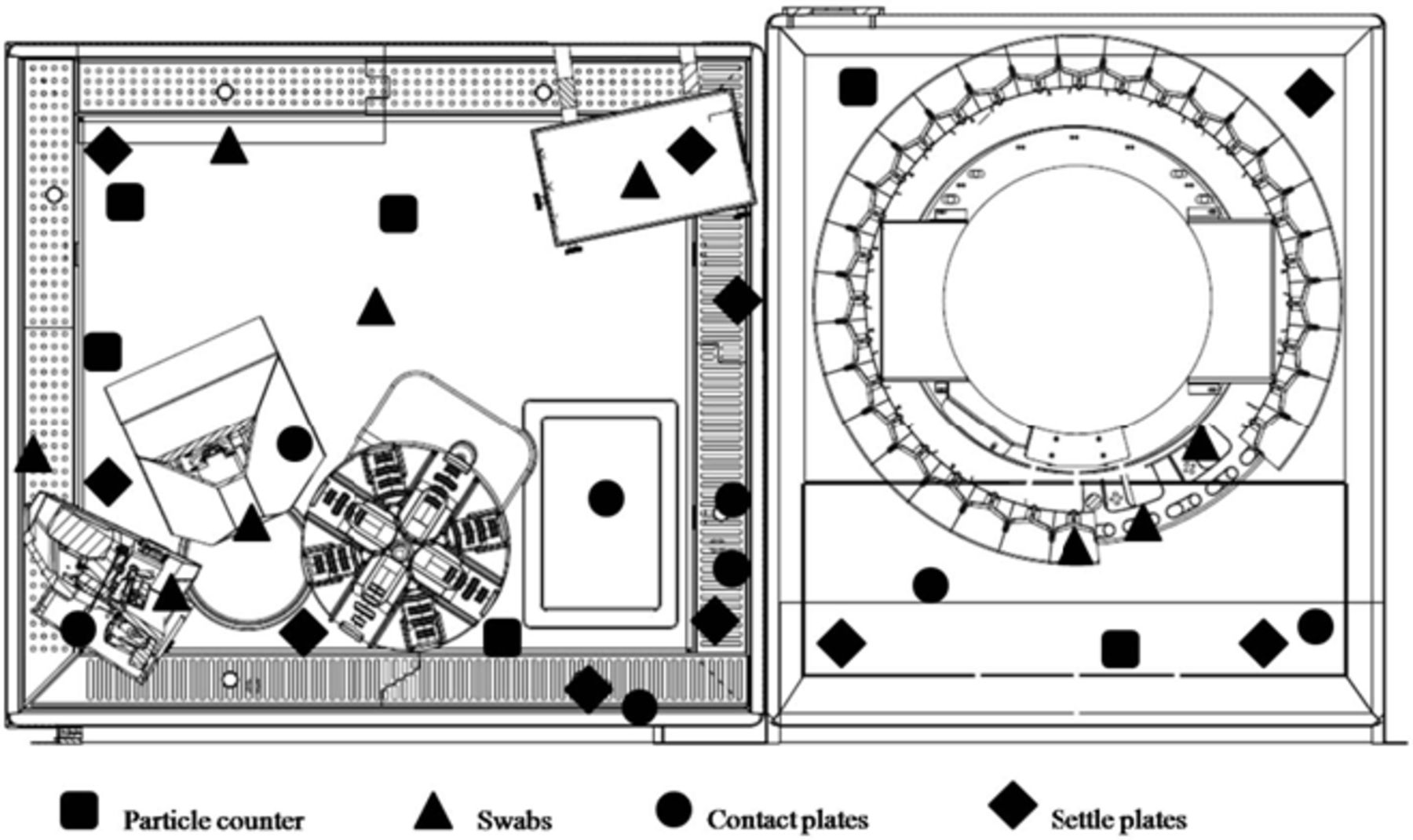
Environmental monitoring sample map.
Efficacy of the automatic microbial decontamination process
UV efficacy was tested using a microbiological challenge test, simulating the real operating conditions: Pseudomonas aeruginosa ATCC 9027, Staphylococcus aureus ATCC 6538, Bacillus subtilis ATCC 6633 spores and Candida albicans ATCC 10231 were selected as microbial targets, due to their diverse resistance to UV radiation.19
Overnight cultures of TSA for bacteria and on Sabouraud Dextrose Agar (SDA) for C. albicans were harvested in sterile 0.85% saline solution. Inoculum densities were spectrophotometrically adjusted to the 0.5 McFarland standard. B. subtilis was allowed to sporulate and the spore suspension was heated at 70°C for 30 min to inactivate vegetative forms. For assessing spore quality, the suspension was examined with a phase-contrast microscope. Spore concentration was determined by plating on TSA serial dilutions in sterile distilled water at 36°C±1°C for 24±2 hours. A titer of 1–5×108 CFU/mL was adequate to achieve the test inoculum. The spore suspension was stored at 2°C–5°C until use.20 21
For each strain, two concentrations were selected: 105 and 107 CFU/mL for P. aeruginosa and S. aureus, and of 105 and 106 CFU/mL for C. albicans. One mL of each suspension was spread on TSA and SDA plates. For B. subtilis endospores, a 10 µL suspension containing 103 CFU/mL and 105 CFU/mL respectively were spotted onto sterile cover slips, afterwards air dried in the dark at 37°C and then put on the surface of plates.
A preliminary challenge test was performed selecting P. aeruginosa among the microorganisms, due to its UV-resistance already detected in previous tests.15 Five plates, with concentrations of 105 CFU/mL and 107 CFU/mL respectively, were located in five main shadow zones inside the equipment for different irradiation times until no microbial growth was recorded for every concentration. After irradiation, the plates were incubated at 37°C for 24 hours – 48 hours and then the microbial loads were evaluated.
Then, plates containing all the test microorganisms were directly exposed to UV lamps (55 cm distance) into the compounding room for irradiation times of 1, 2, 3 and 4 hours. At the end of each irradiation period, bacterial spores were recovered from the cover slips following Lindberg and Horneck’s method and spread on TSA plates.22
Cultures of B. subtilis spores, non-spore-forming bacteria and C. albicans were incubated at 36°C±1°C for 24 hours – 48 hours.
Controls were performed keeping duplicates of irradiated plates in a non-exposed location for the whole irradiation time and then incubated under the same conditions.
Counts were performed as described earlier. Microbial viability was expressed as absolute counts. In addition, inactivation curves were plotted as killing rate (logarithmic ratio between the concentration of microorganisms after and before irradiation) versus time of exposure.
Statistical analysis
All the experiments performed were repeated three times.
To evaluate the difference between the microorganisms, we used a one-way ANOVA model, followed by the Bonferroni post-hoc test. The experiment-wise significance level was fixed at 0.05. The two-sample t test was used to compare B. subtilis and C. albicans. All the analyses were performed using Stata 14.2 SE (Stata corporation, Texas, TX).
The power of the test was 90% (alpha=0.01).
Media Fill test for aseptic processing validation
Media Fill tests were performed in consecutive sessions of the batch productions of both bags and syringes: three runs per day, where each run was composed of 21 bags and eight syringes, repeated over 5 days. A total of 435 Media Fill preparations were compounded. Ready-to-use injection solutions of TSB were used as a medium. Double-strength TSB in 100 mL vials, single-strength TBS in 15 mL vials, 0.9% sodium chloride 50 mL bags (Viaflo, Baxter Healthcare Ltd, Thetford, UK) and sterile 20 mL – 50 mL syringe (Drug Compounding Dosing Device ASN-50, Loccioni, Rosora, IT) were used. Before the materials were loaded in the robot, the phial and bag rubber stoppers were disinfected manually with ethanol (70%) inside the loading area. Positive controls were performed by inoculation of S. aureus, P. aeruginosa and C. albicans solutions inside one TSB phial for each run.
At the end of the batch production, final products were incubated for 7 days at 22.5°C to check the growth of psychrophilic microorganisms: after 7 days at 32.5°C to verify the growth of mesophilic microorganisms.18
Partially used vials unloaded during the cycle followed the same incubation process in order to validate microbiological quality.
Acceptance criterion for an approved sample was no microbial growth by visual inspection.
Media Fill bags were prepared by 50 mL double-strength TSB withdrawal from 100 mL phials and injection in 50 mL-bags, whereas, syringe preparations provided for the withdrawal of 10 mL of single-strength TSB from 15 mL vials in a 20 mL syringe.
The simulation tests comprised the whole production process, including the automatic needle removal from syringes and the screwing of tamper-evident caps (B. Braun Melsungen AG, Melsungen, DE) the automatic labelling and discharge of the final products (figure 4).

Media Fill test. Respectively phial, syringe and bag manipulation.
Results
Qualification of the Class-A inner environment of the robot
The monitoring inside the equipment showed no evidence of microorganism growth in the air and surface samples, at rest before and after production simulation tests, and during compounding.
The particulate matter sampling did not detect any particles from the 0,3 µm to 10 µm diameter range inside the equipment ‘at rest’. No differences were detected between starting and final sampling. In a few instances small particles (0.5 µm) were detected ‘in operation’, but the number of particles were below the limit accepted for the Class A.14 In detail, the maximum value recorded was two particles/m3, while the limit is 3250 particles/m3. No larger particles were ever detected ‘in operation’.
Efficacy of the automatic microbial decontamination process
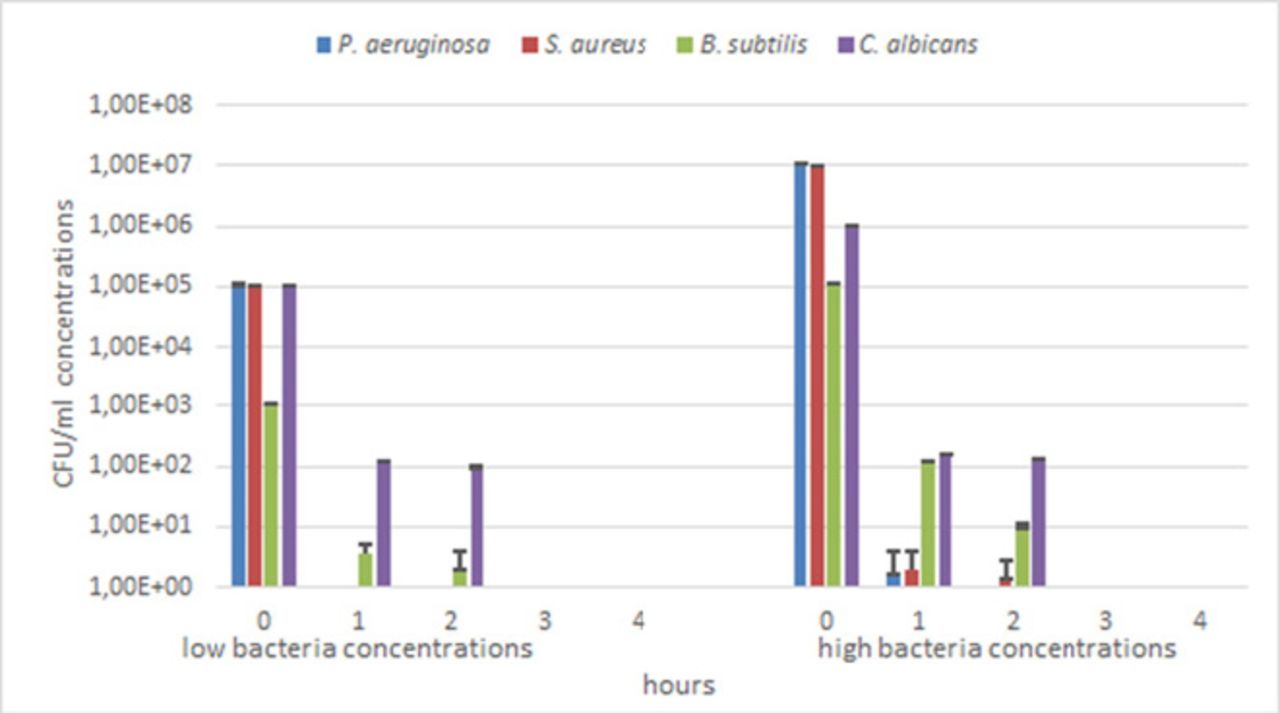
The preliminary test showed that P. aeruginosa did not retain viability after 2 hours. More extensive studies using 107 CFU/mL concentration, only 1–2×100 CFU/mL (about 7-log inactivation) were observed after 1 hour of UV irradiation in all of the selected shadow zones, whereas no colonies were found at the105 CFU/mL concentration after 1 hour of UV irradiation.
After 3 hours of direct UV irradiation, none of the tested microorganisms retained viability (figure 5).

Microbial growth at two concentrations and different UV irradiation times.
For P. aeruginosa and S. aureus, complete inactivation was achieved within the first hour of UV irradiation for the 105 CFU/mL concentration, and within the second and third hours at 107 CFU/mL concentration respectively. In all cases, a significant decrease in viable cells was recorded since the first hour of UV irradiation. At the higher concentrations, after the first hour, for P. aeruginosa and S. aureus, a viable count of 1.67×10°CFU/mL (6.78-log inactivation) and a 2×10°CFU/mL count (6.70-log inactivation) was observed respectively: for S. aureus, 1.34×10°CFU/mL (6.87-log inactivation) were detected also after the second hour. At 103 CFU/mL concentration, B. subtilis spores showed counts of 3.67×100 (2.44-log inactivation) and 2.0×10°CFU/mL (2.70-log inactivation) after 1 and 2 hours of UV irradiation respectively. A reduction of 105-fold (2.94-log inactivation) within the first hour and a further reduction in the order of 10-fold (4.03-log inactivation) in the second hour was registered at 105 CFU/mL concentration. Finally, C. albicans at 105 CFU/mL concentration showed a decrease in viability of the order of 103-fold (2.92-log inactivation) and 104-fold (3.02-log inactivation) within the first and second hour of UV irradiation respectively: at 106 CFU/mL concentration the viability reduction was in the order of 103-fold after both the first and the second hour of UV irradiation with inactivation rates of 3.82-log and 3.88-log respectively (figures 5 and 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Microbial inactivation rate during 2-hour UV irradiation time expressed as the log of the ratio between the viable count after irradiation (Nt) and the viable count before irradiation (N0).
At the highest concentrations, statistical analysis evidenced significant differences between B. subtilis and the other bacteria (P<0.01) and C. albicans (P<0.05) after 1 hour of UV irradiation. At the lower concentrations significant differences were seen only between C. albicans and B. subtilis after 1 (P<0.01) and 2 hours (P<0.05) of UV irradiation.
Media fill test – aseptic processing validation
None of the bags and syringes handled in the simulated production cycle showed turbidity after the incubation period, thereby indicating no microbial contamination. On the contrary, positive controls turned turbid within the incubation period.
Discussion
In the study, we performed an exhaustive investigation into sterile manufacturing and disinfection techniques of the new IV-compounding robot. Parenteral preparations manufacturing requires an aseptic process for all working steps, executed in the so-called clean-areas, in order to avoid any microbial contamination. According to GMP guidelines for clean-areas classification, sterile compounding has be performed in a Class-A environment, whose recommended limits for microbial contamination are: <1 CFU/ in air sample; <1 CFU/4 hours in settle plates (diameter 90 mm): and <1 CFU/plate in contact plates (diameter 55 mm).14 In parallel, the detection of airborne particulate matter is important to ensure a suitable environment for the preparation of aseptically manufactured products. In fact, particles can enter the products as extraneous contaminants and can act as a vehicle for microorganisms. Standards for Class-A rooms state that maximum values allowed for airborne particulate matter inside the equipment at rest are 3520/m3 and 20/m3 for particles of 0.5 µm and 5 µm diameter, respectively. Our results demonstrate IV-robot compliance with the Class-A requirements for both microbial and particle monitoring.
in air sample; <1 CFU/4 hours in settle plates (diameter 90 mm): and <1 CFU/plate in contact plates (diameter 55 mm).14 In parallel, the detection of airborne particulate matter is important to ensure a suitable environment for the preparation of aseptically manufactured products. In fact, particles can enter the products as extraneous contaminants and can act as a vehicle for microorganisms. Standards for Class-A rooms state that maximum values allowed for airborne particulate matter inside the equipment at rest are 3520/m3 and 20/m3 for particles of 0.5 µm and 5 µm diameter, respectively. Our results demonstrate IV-robot compliance with the Class-A requirements for both microbial and particle monitoring.
The assessment of the automatic microbial decontamination revealed a very effective UV plant design, showing inactivation of high microbial concentration just after 2-hour exposition, also in shadow zones, probably thanks to the joint action of the UVC-generated ozone and of the mirror effect of the steel surfaces inside the equipment. P. aeruginosa and S. aureus turned out to be the most sensitive, showing no viability at 105 CFU/mL concentration after the first hour of exposition, and very high inactivation rates at 107 CFU/mL concentration. On the contrary, C. albicans was the most resistant microorganism, surviving until the second-hour exposition at both concentrations. This is in accord with several authors who report that fungi are more resistant than vegetative bacteria to UV inactivation.23 24 Inactivation of B. subtilis endospores were higher than that of C. albicans. These results confirmed our previous studies,15 although other studies have shown that bacterial endospores are more resistant than yeasts to UV inactivation.25
The complete absence of microbial contaminants found during the environmental qualification of the internal areas of the equipment is an indirect confirmation of the efficacy of the UV irradiation to maintain the aseptic state under ordinary conditions.
Finally, the Media Fill test showed that no samples were contaminated, which supports the high efficiency in sterile manufacturing of the equipment, as a further confirmation of the investigated aseptic operativng conditions and cleaning procedures.
Conclusion
On the basis of these studies, we conclude that the IV compounding robot, APOTECAunit, meets the requirements for sterile compounding, set by ISO14644-1 and EU GMP standards for the hospital pharmacy and the pharmaceutical industry in general. The total absence of growth recorded during the Media Fill test and the environmental qualification indicates that robotics provides advantages in terms of safety for the patient compared with conventional procedures of parenteral preparation production thanks to the high standardisation introduced. In addition, these results suggest that robotic compounding may represent a powerful tool to evaluate the possibility of extending the microbial stability of the final products.
This protocol has demonstrated to be a comprehensive and valuable tool to validate, from a microbial point of view, a sterile-compounding technology. As more and more technologies are implemented in sterile hospital practice, this study might represent an important benchmark to develop a clinic validation protocol, as required, for instance, in the Performance Qualification of the GMP in the case of drug manufacturing.
What this paper adds
What is already known on this subject
Manual compounding is still the most common practice for the preparation of non-hazardous IV medications
Manual IV compounding, performed either by a technician in the hospital pharmacy or by a nurse in the hospital wards, can lead to incorrect doses of medication or microbiological contamination.
The presence of IV compounding robots in hospitals has safety, quality control and efficiency.
What this study adds
This study has shown that the APOTECAunit meets the requirements for advanced aseptic processing in the hospital pharmacies, provides advantages in terms of safety for patients compared with conventional procedures of parenteral preparation production.
This study provides a method to develop a contamination control strategy to evaluate sterility, as required in the Performance Qualification of the GMP for drug compounding.
Robotic compounding represents a powerful tool to extend the sterility of the final product.
References
Footnotes
EAHP Statement 5: Patient Safety and Quality Assurance.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.